
Druggability Assessment Services: A Practical Guide for Drug Discovery Teams
A structured resource for biotech and pharma teams navigating early-stage drug discovery decisions — by Rinat Borenshtain-Koreh, PhD, DVM
Expert Perspective — 25+ Years in Biotech R&D
Most molecules that look promising in a screening campaign will never reach the clinic. The reasons are rarely about raw potency — they are about absorption, metabolism, target accessibility, and dozens of physicochemical realities that only surface under rigorous, structured testing. As a scientist with over 25 years designing biological models, in-vitro assays, and in-vivo experiments for medical and biotech R&D — including FDA application support — I can state clearly: structured druggability assessment before budget lock-in is the single most cost-effective decision a drug discovery team can make.
⚡ Exclusive Insight
The cost of pursuing an undruggable target is not just money — it is the opportunity cost of every program that was not funded instead. Teams that integrate structured druggability assessment at the Hit-to-Lead stage consistently retire fatal risks before they become fatal expenses. A clear, data-driven evaluation is not a gatekeeping exercise; it is the fastest route to a confident Go decision.
What Are Druggability Assessment Services and Who Are They For?
Druggability assessment services are a structured set of biological, biochemical, and physicochemical evaluations designed to answer one question early: does this target — and this molecule — have a realistic path toward becoming a drug? The work is most valuable during Target ID/Validation and Hit-to-Lead, where decisions still cost relatively little.
Teams that integrate Da-Ta Biotech R&D services at this stage can map risks before scale-up commitments. The cost of pursuing an undruggable target is not just money; it is the opportunity cost of every program that was not funded instead. A clear, data-driven assessment prevents the classic sunk-cost trap of doubling down on a candidate that the data is already warning against.
🎯 Target ID / Validation
Identify whether your target has accessible binding sites and biological relevance before committing to a scaffold.
⚗️ Hit-to-Lead
Filter screening hits by biological and physicochemical criteria so chemistry optimization focuses on viable candidates.
📊 Lead Optimization
Confirm that medicinal chemistry improvements translate into measurable gains across ADME, selectivity, and potency.
How Do You Know If a Molecule Is a Drug?
The question Is this molecule a drug? is deceptively simple. Potency in a primary assay tells you almost nothing about whether the molecule can survive the body, reach its target, and act selectively. Drug-like properties testing combines biological efficacy with physicochemical evaluation — solubility, permeability, plasma stability, and metabolic behavior — so that “active” does not get confused with “developable.”
A compound can be exquisitely potent and still fail because it precipitates in gastric fluid or vanishes in the first metabolic pass. This is not a theoretical concern — it is the most common failure mode in modern drug discovery.
Early Warning Signs That a Lead Molecule May Fail
Common red flags include the following. None are automatic disqualifiers, but each one signals that medicinal chemistry will need to invest in fixing a property rather than optimizing efficacy. Catching them early is the entire point of the exercise.
⚠️ Red Flags in Lead Molecules
- Poor aqueous solubility — below approximately 10 µg/mL
- High lipophilicity — LogP greater than 5
- Rapid microsomal turnover indicating fast first-pass metabolism
- Strong CYP inhibition creating drug-drug interaction risk
- Unexpected plasma protein binding profiles reducing free fraction
Three Terms People Confuse: Druggability, Drug-Likeness, and Developability
These words get used interchangeably, and the confusion costs programs real time. Understanding the distinction is foundational to designing the right assessment strategy.
Druggability
Target-Centric
Does the protein have accessible hot spots — pockets with the right geometry, hydrophobicity, and dynamics to bind a small molecule with useful affinity and selectivity?
Drug-Likeness
Compound-Centric
Does the molecule itself sit in a chemical space consistent with oral bioavailability, metabolic survival, and the physicochemical properties of known drugs?
Developability
Product-Centric
Can the molecule be manufactured, formulated, and stored as a viable pharmaceutical product? This includes scalability, excipient compatibility, and shelf-life.
Mixing these up leads teams to fix the wrong problem. Drug-like properties testing addresses the second question; structural and biophysical analysis addresses the first. A great target with a bad compound is still a fixable problem. A bad target with a great compound rarely is.
Step-by-Step: What a Professional Druggability Evaluation Entails
A robust druggability evaluation moves through defined phases. The goal is not to generate data for its own sake — it is to retire specific risks in a defined order, with each experiment carrying a clear hypothesis and a clear cost.
Data Collection
Published literature, structural data, prior assay results, and target biology are compiled into a unified evidence base.
Target Characterization
Is the binding pocket well-defined? Are there validated chemical starting points? What is the target’s biological validation status?
Target Product Profile
A TPP describing the desired final product attributes — dose form, potency range, selectivity requirements — anchors all subsequent decisions.
Setting Success Criteria and Data-Driven Decision Making
Predefined pass/fail metrics are non-negotiable. Decide before the experiment what solubility threshold, what IC50 range, or what stability half-life will trigger a Go/No-Go. Without these, results get rationalized after the fact, and weak candidates survive longer than they should. This is not scientific conservatism — it is the standard practice of every well-run discovery program.
🔧 Methodology: De-Risking Plan Construction
A de-risking plan is constructed as the final step — a roadmap of experiments, each with a clear hypothesis, a clear cost, and a clear decision node. The plan answers: what would we need to see to keep investing in this candidate, and what would we need to see to stop?
- Each experiment maps to a specific risk category
- Results are tied to actionable next steps, not just data generation
- Resources are allocated sequentially — highest-risk questions first
Why Early ADME Is Essential for Drug-Like Properties Testing
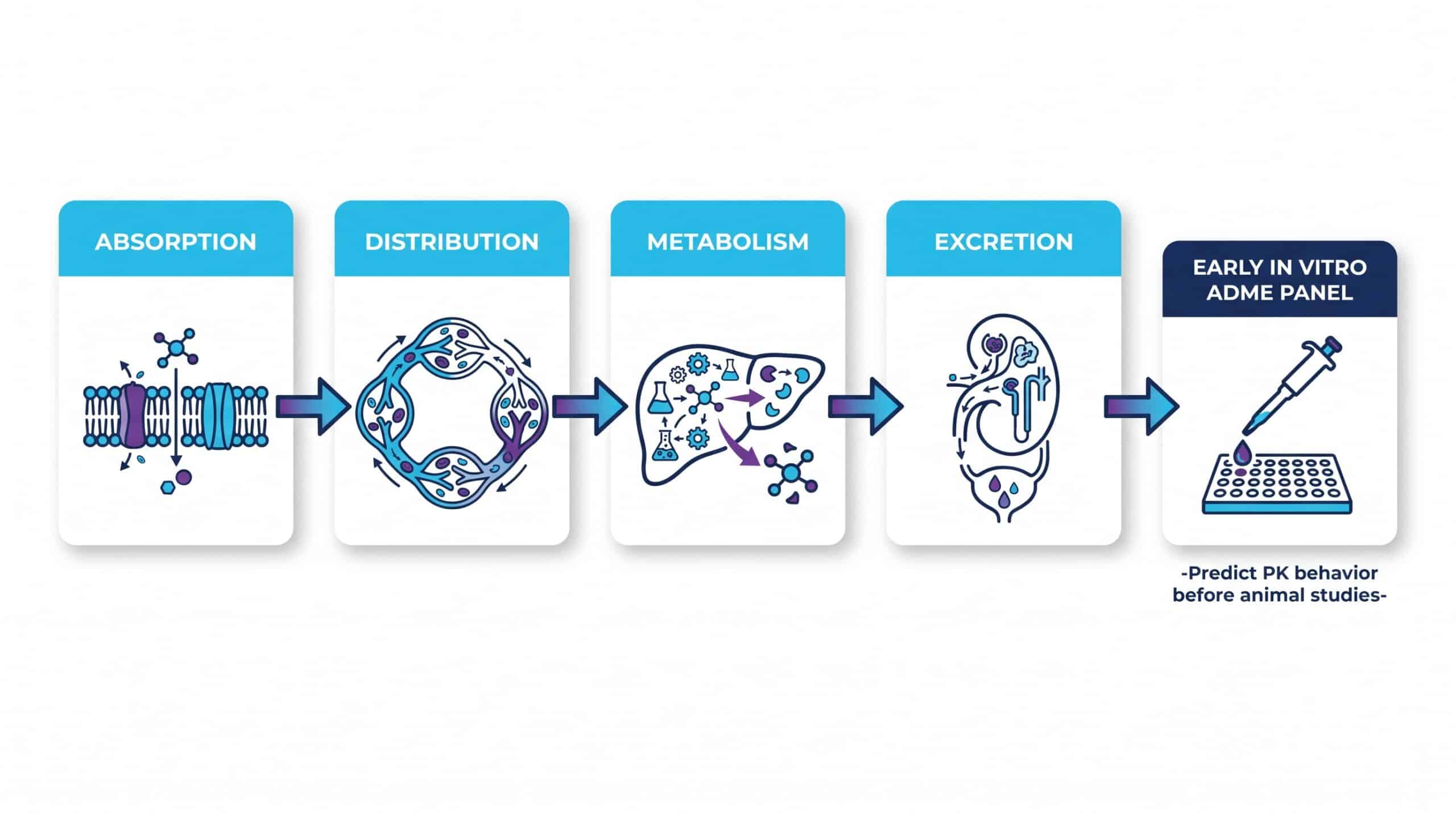
ADME — Absorption, Distribution, Metabolism, Excretion — is where many late-stage failures originate. Running early in vitro ADME panels lets teams predict pharmacokinetic behavior before committing to animal studies or expensive optimization cycles. Regulatory bodies have made this expectation explicit; the FDA’s guidance on in vitro metabolism and transporter-mediated DDI studies describes the standard expectations for risk assessment.
Skipping these steps does not save time — it relocates the failure to a more expensive phase. Every week invested in early ADME characterization can prevent months of misdirected lead optimization.
The Basic Essential ADME Test Panel
A foundational panel typically includes the following. These tests are inexpensive relative to what they prevent:
✅ Core ADME Panel Components
- Kinetic and thermodynamic solubility
- LogD/LogP measurement (lipophilicity profiling)
- Plasma and microsomal stability
- Permeability — Caco-2 or PAMPA
- Basic CYP inhibition screening (CYP3A4, CYP2D6, CYP2C9)
Target Druggability Assessment vs. Compound Assessment
These are two different exercises that often get bundled together. Target druggability asks whether the protein has accessible “hot spots” — pockets with the right geometry, hydrophobicity, and dynamics to bind a small molecule with drug-like affinity. The foundational framework for this kind of druggability classification remains widely cited in the literature.
Compound assessment, in contrast, evaluates the chemical viability of a specific candidate: its synthesis tractability, its physical behavior in solution, and its metabolic profile in relevant biological matrices.
🎯 Target Druggability
- Structural pocket analysis
- Binding site geometry & dynamics
- Biophysical confirmation (SPR, NMR)
- Target biological validation
⚗️ Compound Assessment
- Physicochemical profiling
- In vitro ADME panel
- Synthetic accessibility & scalability
- Selectivity over related targets
“A great target with a bad compound is still a fixable problem. A bad target with a great compound rarely is. The distinction is not semantic — it determines where medicinal chemistry resources should be spent.”
— Rinat Borenshtain-Koreh, PhD, DVM — Da-Ta Biotech LTD
What Makes a High-Potential Druggability Compound
A druggability compound is one that balances potency with physical and metabolic stability. High affinity is meaningless if the compound aggregates in solution, binds promiscuously to plasma proteins, or is cleared within minutes of administration.
The traits to look for — and the ones a well-designed assessment will measure — include:
Moderate MW
Typically under 500 Da for orally bioavailable small molecules
Aqueous Solubility
Measurable solubility at physiological pH — above 10 µg/mL as a baseline
Defined Selectivity
Confirmed selectivity over related or off-target proteins in the same family
Do General “Rules of Thumb” Guarantee a Successful Drug-Like Molecule?
Lipinski’s Rule of Five and similar heuristics are useful baselines, but they are not predictive in the strict sense. Many marketed drugs violate one or more rules; many rule-compliant compounds fail in development. The rules describe statistical tendencies in historic oral drugs — not physical laws.
⚠️ Critical Reminder
Treat rules of thumb as a starting filter, not as a substitute for empirical testing. A molecule that passes every rule but precipitates at physiological pH is still not a drug. Computational compliance is not the same as physical reality. Rigorous in vitro testing remains the only way to confirm what rules can only estimate.
In Silico vs. In Vitro Druggability Assessment — Which Is Better?

This framing creates a false choice. In silico methods deliver speed and breadth: structural pocket analysis, docking, ADME prediction, and cheminformatics filters can screen thousands of compounds in hours. In vitro work delivers physical truth: actual solubility numbers, actual metabolic half-lives, actual binding affinities in a relevant biochemical environment.
The right druggability evaluation uses computational triage to prioritize, then uses bench experiments to verify. Computational predictions that have not been physically tested are hypotheses, not data — and no investor, regulatory reviewer, or development team should treat them as anything more.
“Computational predictions that have not been physically tested are hypotheses, not data. A robust assessment integrates both — using computational triage to prioritize and bench experiments to confirm.”
— Rinat Borenshtain-Koreh, PhD, DVM — Da-Ta Biotech LTD
Comparison Table: Assessment Approaches at a Glance
Each approach answers a different question and carries different costs and limitations. A well-designed program uses all four in sequence, not as alternatives to one another.
Key Deliverables of a Druggability Assessment Service
A useful service produces three things: a comprehensive risk report that names each weakness explicitly, raw and processed data the team can re-analyze independently, and actionable recommendations for the next development phase. The report should not read like a press release. It should identify which risks are likely showstoppers, which can be engineered around, and which require additional experiments before a responsible Go/No-Go can be made.
Mapping Business Needs to Assessment Outputs
Time, Budget & Common Mistakes in Druggability Work
How Much Time and Budget Should You Allocate?
Costs scale with the depth of the assessment. A focused early-stage panel — solubility, basic stability, primary potency confirmation — can be completed in two to four weeks at modest cost. A full druggability profile that includes biophysical confirmation, ADME, and cell-based functional work runs longer, typically two to three months.
The modular structure matters: teams can start narrow and expand only when the data justifies further investment. Spending more on a candidate before basic risks are retired is a common and avoidable mistake.
When to Initiate a Druggability Evaluation
The honest answer is: as early as your data allows. Once a target is selected and initial hits are identified, druggability questions should already be on the table. Waiting until lead optimization means accumulating sunk costs around assumptions that have never been tested. Early evaluation is not premature — it is protective.
Common Mistakes Teams Make in Druggability Work
Several patterns recur across programs. Each is correctable, but only if the team treats druggability as a discipline rather than a checkbox.
⚠️ Recurring Mistakes to Avoid
- Over-relying on a single potency assay and skipping orthogonal confirmation
- Running ADME too late, after chemistry has locked in a scaffold
- Interpreting rule-of-five compliance as a development green light
- Commissioning expensive cell work before confirming basic biochemical activity
- Accepting vendor reports without examining the underlying raw data
Quality Benchmarks & Choosing the Right Partner
What to Verify in Any Assessment
How to Choose the Right Partner for Druggability Assessment Services
Selecting a biotech R&D partner deserves the same rigor as selecting a candidate molecule. Look for transparency in methodology — a partner who can explain exactly how each assay is run and what its limitations are. Look for clear deliverables defined upfront, not vague promises. Look for proven experience across cell-based, biochemical, and molecular assays.
And most critically: look for a partner willing to tell you when the data is bad news. A vendor who only delivers good news is not delivering science — they are delivering comfort at the cost of your program’s future.
✅ Da-Ta Biotech Quality Assurance
- ISO 9001:2015 quality management framework
- On-site facilities at Rehovot Science Park, Israel
- 25+ years across cell-based, biochemical, and molecular assays
- FDA application support experience for IND-enabling pathways
- Ethical collaboration with IRB/IACUC oversight for in vivo components
Frequently Asked Questions
Summary and Look Forward
Drug development is a marathon measured in years, not quarters, and proper validation early in the race is the best insurance policy a program can carry. Structured druggability assessment turns expensive late-stage surprises into cheap early-stage decisions. The teams that build this discipline into their workflow consistently move faster than the teams that skip it — not because they take shortcuts, but because they stop investing in candidates that the data was already warning against.
Are your current candidates being evaluated against the right physicochemical and biological criteria, or are you optimizing potency in a vacuum? If you want a clear-eyed look at what your molecule can and cannot become, our team can help you design the right experimental path.
Explore testing drug-like properties with us, or reach out through our contact page to start the conversation. Come to us with any scientific challenge — we are here for you.

This article is intended for scientific and educational purposes. All assessment data referenced reflects general industry standards. For program-specific advice, consult directly with a qualified R&D partner.
