
Drug Safety Testing In Vitro: A Practical Guide for R&D Decision-Makers
✦ Written by a Scientific Authority with 25+ Years in Biotech & Biomed R&D
With direct experience in biological model development, in-vitro assay design, IND-enabling studies, and FDA application support, this guide translates complex preclinical methodology into clear, actionable decisions for R&D leaders. In vitro safety data is not optional — it is the first filter that separates viable candidates from costly failures.
🔬 Expert Insight
Running cytotoxicity, basic ADME, and hERG screens during hit-to-lead optimization is among the highest-ROI activities in drug discovery. Every red flag identified in vitro eliminates months of wasted animal studies and tens of thousands in avoidable expenditure. The earlier, the cheaper — and the smarter.
Why In Vitro Safety Is the First Filter in Drug Discovery
Drug safety testing in vitro is the initial gatekeeper of the pharmaceutical pipeline. Before a candidate molecule sees an animal or a human, it must demonstrate that it does not collapse cell viability, disrupt critical organ function, or generate harmful metabolites. Cell cultures and biochemical assays serve as controlled environments to simulate human biological responses — fast, focused, and far less costly than late-stage failure.
According to the FDA’s definition of preclinical research, sponsors are required to conduct both in vitro and in vivo studies to evaluate toxicity before advancing to clinical trials. This dual requirement positions in vitro work as a foundational pillar of the broader preclinical safety assessment framework — not a shortcut, but a scientifically grounded triage tool.
From Animal Models to Animal-Free: A Shift in Methodology
Modern R&D is moving steadily toward “animal-free” methodologies. This is not just an ethical evolution — it is a scientific upgrade. Human-derived cell systems (primary hepatocytes, iPSC-cardiomyocytes, organoids) often predict human responses more reliably than rodent models for certain endpoints. Regulatory bodies increasingly accept validated in vitro data as part of integrated risk assessments, particularly when paired with mechanistic insight.
This is exactly where early toxicological evaluation services add value: they help research teams narrow down lead compounds by exposing safety liabilities before resources are committed to advanced development. The earlier a red flag appears, the cheaper it is to act on it.
What Does a Standard Drug Toxicity Test Include?
A drug toxicity test is designed to surface the red flags — cell death, membrane rupture, mitochondrial dysfunction, or impaired proliferation. The cornerstone is cytotoxicity, which measures the compound’s effect on cell viability, membrane integrity, and metabolic activity. Common readouts include MTT/MTS, LDH release, ATP quantification, and live/dead imaging.
Beyond general cytotoxicity, organ-specific toxicity is critical. Hepatotoxicity and cardiotoxicity remain the leading causes of clinical and post-market drug failure, and they are best probed early using cell-based assays on relevant lineages such as HepG2, primary hepatocytes, or hiPSC-derived cardiomyocytes.
Cytotoxicity Readouts
- MTT / MTS assay
- LDH release
- ATP quantification
- Live/dead imaging
Organ-Specific Cell Lines
- HepG2 hepatocytes
- Primary hepatocytes
- hiPSC-derived cardiomyocytes
- Relevant human-derived organoids
Liver Toxicity: The Most Common Offender
Roughly a third of drugs fail clinical studies due to safety concerns, and the most common issue is liver toxicity. This is why hepatocyte-based assays — covering viability, metabolic competence, and bile acid handling — sit at the center of any serious early safety panel. Detecting hepatic liabilities in vitro saves programs from collapsing in Phase I or II.
⚠️ Warning: Don’t Skip Hepatic Profiling
Hepatotoxicity discovered after Phase II is exponentially more costly than the same finding at lead optimization. A single hepatocyte assay panel — viability, CYP activity, and transporter function — can flag the majority of clinically relevant liver liabilities.
The Critical Role of ADME Tox Testing
ADME Tox testing — Absorption, Distribution, Metabolism, Excretion, and Toxicity — provides the systemic context that pure cytotoxicity cannot. Looking at “Tox” in isolation is insufficient. A molecule may appear clean in a viability assay, yet generate reactive metabolites once metabolized by the liver. The toxic species is often not the parent drug.
ADME studies determine the “drug-likeness” of a molecule and its potential for Drug-Drug Interactions (DDI). At Da-Ta Biotech, ADME-Tox is treated as an integrated profile rather than a checklist — connecting metabolic stability, transporter behavior, and toxicity into one decision-grade dataset.
✅ Best Practice: Integrate, Don’t Isolate
Treat ADME and Tox as one unified dataset. Metabolic stability data must inform toxicity interpretation — a rapidly cleared molecule with a toxic metabolite is a fundamentally different risk than a stable compound with direct cytotoxic activity.
CYP Enzymes and Transporters: Predicting Interactions
The cytochrome P-450 (CYP) enzyme family metabolizes the majority of small-molecule drugs. CYP inhibition or induction can dramatically alter the plasma levels of co-administered medications, creating safety risks. Transporter-mediated interactions add another layer. The reference document on in vitro CYP and transporter-mediated drug interaction studies outlines why these assays are now standard in any IND-enabling package.
“CYP inhibition or induction data generated in vitro are used to predict the potential for drug-drug interactions in vivo, often obviating the need for dedicated clinical DDI studies when the in vitro signal is clearly negative.”
— ICH / FDA Guidance on Drug Interaction Studies
Key Assays and Endpoints: How Do You Actually Measure Safety?

An effective safety panel combines multiple assays, each targeting a distinct biological liability. Endpoints — the measurable outcomes such as IC50 values, percentage of inhibition, or fold-induction — are what translate raw signal into actionable decisions.
The ICH S7B guideline defines how hERG and related assays are used to evaluate delayed ventricular repolarization — a non-negotiable element of cardiovascular safety assessment.
Common Mistakes Teams Make in Early Safety Screens
Several recurring errors weaken otherwise well-designed studies. Each mistake quietly inflates false-negative rates — the most expensive kind of error in drug development.
❌ Testing Only Parent Compound
Without metabolic activation, you miss the actual toxic species. The parent molecule is often not what damages tissue — its metabolite is.
❌ Ignoring DMSO Solvent Effects
DMSO at even 0.5% can alter baseline cell viability. Controls must account for solvent concentration at every test point.
❌ Single Cell Line as “Human” Proxy
One cell line cannot represent whole-body safety. A multi-lineage panel covering liver, heart, and kidney is the minimum credible screen.
❌ Missing Concentration-Response Curves
Reporting a single concentration endpoint has no scientific validity for go/no-go decisions. IC50 or EC50 derivation requires a properly designed concentration-response experiment.
A Scenario: When In Vitro Data Saves a Program
Case Study: Kinase Inhibitor Rescued Before Animal Studies
A small-molecule kinase inhibitor passes basic cytotoxicity screening in HepG2 cells — initial signal looks clean. The team proceeds to metabolic stability testing.
- Finding 1: Rapid metabolic turnover detected in liver microsomes — intrinsic clearance too high for viable dosing.
- Finding 2: CYP3A4 induction detected — significant DDI risk flagged.
- Finding 3: Follow-up hepatocyte assay reveals reactive metabolite formation.
- Outcome: Team reformulates a backup analog before any animal study is initiated — months saved, budget protected.
This is the practical value of integrated ADME Tox testing: not just data, but strategic direction for the program.
Choosing a CRO: What to Look for in Preclinical Safety Assessment
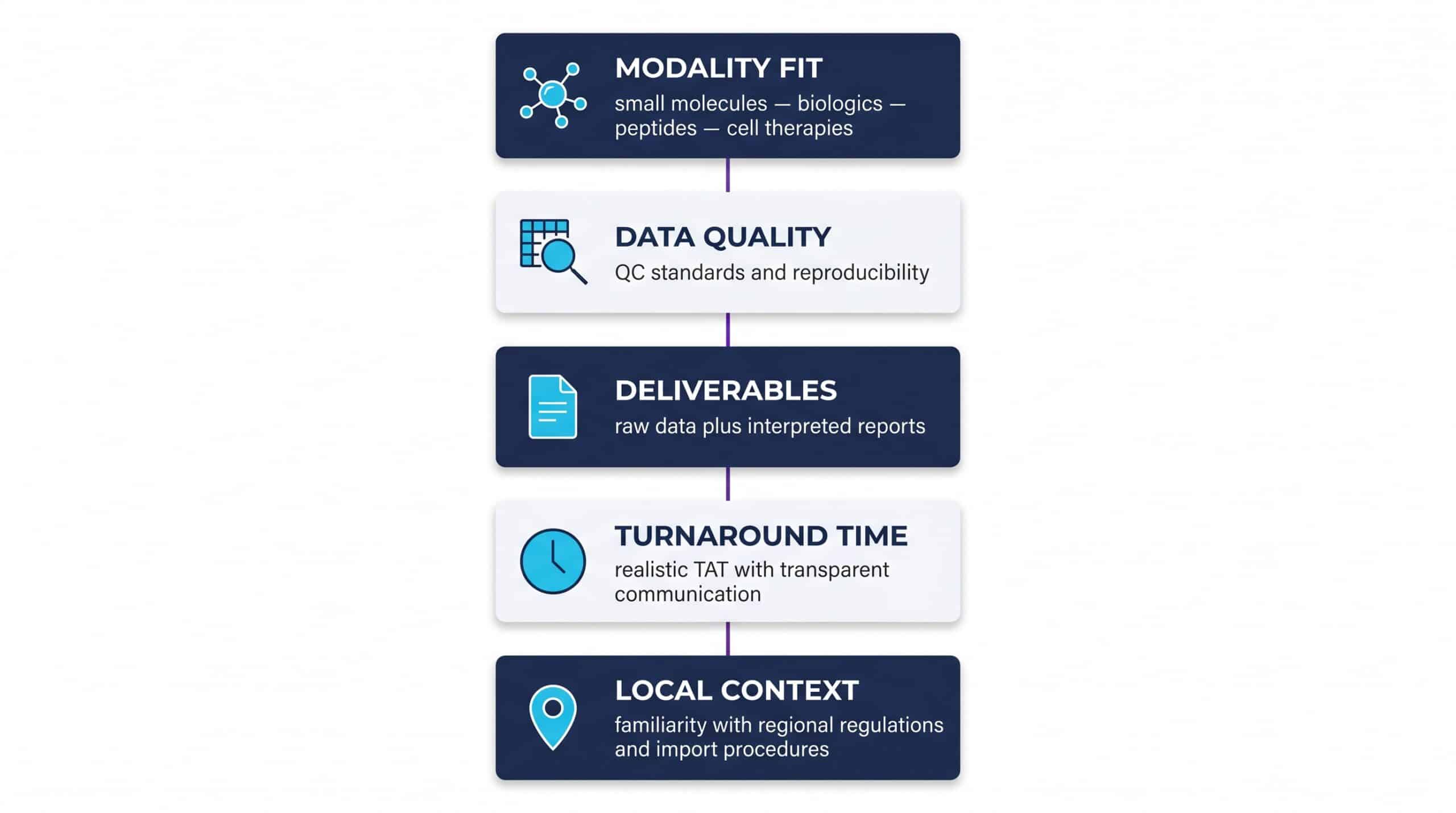
Selecting a laboratory partner for preclinical safety assessment is a decision that shapes the entire trajectory of a program. Speed matters, but reproducibility, modality coverage, and clarity of deliverables matter more.
For Israel-based programs, regional expertise simplifies navigation of Ministry of Health import procedures for research compounds. A partner located in the Israeli science ecosystem — such as Da-Ta Biotech in Rehovot’s science park — combines proximity, ISO 9001:2015 quality standards, and personalized scientific consulting tailored to early-stage R&D teams.
How Da-Ta Biotech Supports Decision-Making
Beyond running assays, the team functions as a scientific partner from proof-of-concept through demonstrations of efficacy. That means flexibility in protocol design, willingness to engage with a customer’s specific molecule and biology, and reports that translate endpoints into go/no-go decisions.
🏆 What Researchers Say About Working with Da-Ta Biotech
- Robust, well-established cellular models and validated protocols
- ISO 9001:2015 quality management standards
- Reports that interpret endpoints — not just deliver raw numbers
- “We are here for you” working style that adapts to each program’s science
“Profiling the druggability and safety of your molecule with validated, well-established protocols is not optional — it is the correct thing to do. Come to us with any scientific challenge.”
— Rinat Borenshtain-Koreh, PhD, DVM | CEO, Da-Ta Biotech LTD
🔬 Da-Ta Biotech Methodology: Integrated Safety Profiling
Our preclinical safety assessment workflow follows a structured, tiered approach designed to maximize information yield per compound at the lowest resource cost:
- Tier 1 — Primary Screen: Cytotoxicity in 2–3 relevant human cell lines at 8–10 concentration points. Establishes basic viability profile and EC50.
- Tier 2 — ADME Panel: Metabolic stability (HLM + hepatocytes), Caco-2 permeability, plasma protein binding, and aqueous solubility.
- Tier 3 — Mechanistic Safety: CYP inhibition (5–7 isoforms), hERG electrophysiology, reactive metabolite trapping, and mitochondrial toxicity.
- Tier 4 — Integrated Report: Data synthesis connecting ADME and Tox findings into a single risk-stratified decision document for program leadership.
FAQ — Common Questions About In Vitro Drug Safety Testing
Have a Molecule That Needs Safety Profiling?
Are you weighing the right balance between speed, depth, and reliability for your next safety study? Da-Ta Biotech is ready to design the in vitro panel your program actually needs — with validated protocols, expert interpretation, and a scientific partner mindset from day one.

