
Drug Development Pipeline Stages: A Practical Guide from Discovery to IND Filing
Bringing a new drug to market is not a single project — it is a pipeline. A continuous, multi-stage journey where scientific rigor, regulatory strategy, and manufacturing readiness must converge at every decision point. From the earliest spark of target identification through formal preclinical drug development and into clinical trials, each stage carries its own risks, milestones, and gate decisions that determine whether a molecule moves forward or stops.
This guide was authored by Dr. Rinat Borenshtain-Koreh, CEO of Da-Ta Biotech LTD, with over 25 years of hands-on experience in biological model development, in-vitro assays, in-vivo experiments, and FDA application support. Every insight here is grounded in real pipeline execution — not theory.
The single most common reason drug programs fail is not poor science — it is misaligned timelines between preclinical data generation, CMC development, and regulatory strategy. Programs that integrate these three workstreams from candidate selection onward consistently outperform those that treat them sequentially. This guide maps exactly when and why each discipline must engage.
This guide provides a practical, end-to-end overview of the drug development pipeline stages. It covers the drug discovery pipeline, the transition into IND-enabling studies, and the cross-functional disciplines — CMC, regulatory affairs, quality — that run parallel to every phase. Whether you are a researcher at an academic institution, a scientist at an early-stage biotech, or a decision-maker evaluating your pharma development process, this resource maps out what happens, when, and why it matters.
What Are the Drug Development Pipeline Stages (In Simple Terms)?
The drug development pipeline stages describe the structured journey a potential therapeutic compound takes from an initial idea to an approved medicine available to patients. While the terminology can vary between organizations, the standard phases are widely recognized: Discovery, Preclinical, Clinical (Phase 1, Phase 2, Phase 3), Regulatory Approval, and Post-Market Surveillance (Phase 4).
Each stage has defined objectives. Discovery focuses on finding the right molecule. Preclinical testing evaluates safety in non-human models. Clinical trials assess safety and efficacy in humans, progressively expanding the patient population. Regulatory review examines the totality of evidence. Post-market surveillance monitors real-world performance. These stages are distinct, yet many activities — regulatory planning, manufacturing development, quality assurance — span across multiple stages simultaneously.
Stages vs. Cross-Functional Disciplines (CMC, Regulatory, Quality, Biostatistics)
It is tempting to view drug development as purely sequential: finish one stage, then start the next. In practice, cross-functional disciplines operate continuously throughout the pipeline. Chemistry, Manufacturing, and Controls (CMC) begins during lead optimization and extends through commercial manufacturing. Regulatory affairs teams plan submission strategies years before an actual filing. Quality assurance establishes protocols from the first GLP study onward. Biostatisticians design clinical endpoints during preclinical phases.
Ignoring parallel workstreams is one of the most common reasons programs experience costly delays. A molecule with promising efficacy data but no scalable manufacturing process cannot advance. Robust cross-functional integration from day one is essential for successful progression through the drug development pipeline stages.
What Is the Difference Between Drug Discovery Pipeline and Drug Development Process?
These two terms are often used interchangeably, but they refer to distinct scopes within the overall journey. The drug discovery pipeline focuses on identifying and optimizing a potential therapeutic compound — a “candidate drug.” This phase involves target identification, hit finding, hit-to-lead progression, and lead optimization. The output is a well-characterized molecule ready for formal testing.
The drug development process, by contrast, encompasses everything from candidate selection through preclinical drug development, clinical trials, regulatory submission, approval, and post-market monitoring. It is the translational bridge between a laboratory compound and a medicine that reaches patients. Understanding this distinction matters because the skill sets, regulatory requirements, and financial investments differ significantly between the two phases. As described in a comprehensive overview by Appsilon, the development process involves rigorous phases, timelines, and key stages that require specialized expertise at each step.
Why Is Da-Ta Biotech a Key Partner in Both Phases?
The transition from discovery to early development is where many programs face their greatest challenges. In vitro biological assays, cell-based models, and ADME profiling are critical in both phases — during discovery to characterize hits and leads, and during early development to generate the data packages required for regulatory submissions. Companies like Da-Ta Biotech offer essential R&D services, supporting crucial activities from the drug discovery pipeline through early preclinical stages, making them a valuable partner in the overall pharma development process. Their ISO 9001:2015-certified laboratory provides validated biological assays that serve as reliable tools for researchers navigating these critical transitions.
Why Is It Called a “Pipeline” and Not a “Project”?
The word “pipeline” reflects a reality that a single “project” does not capture: pharmaceutical companies typically advance multiple drug candidates simultaneously, each at a different stage of development. This continuous flow is a strategic necessity. The attrition rate in drug development is high — estimates suggest that fewer than 10% of candidates entering clinical trials will ultimately receive approval.
Maintaining a pipeline rather than betting on a single molecule allows organizations to diversify risk, allocate resources across programs at varying maturity levels, and sustain a continuous product stream. At each stage transition, teams apply “gate decisions” — go/no-go assessments based on predefined scientific, regulatory, and commercial criteria. A candidate that fails to meet its gate criteria is deprioritized or terminated, freeing resources for more promising molecules. This disciplined approach is what separates successful pharmaceutical organizations from those that overcommit to a single bet.
“A pipeline is not just a portfolio of molecules — it is a portfolio of disciplined decisions. Every gate is an opportunity to protect the whole by being honest about the parts.”
— Dr. Rinat Borenshtain-Koreh, PhD, DVM | CEO, Da-Ta Biotech LTD
What Happens in the Target Identification and Validation Stage?

This is where the drug discovery pipeline begins. Researchers pinpoint specific biological targets — proteins, enzymes, receptors, or signaling pathways — believed to play a causal or contributory role in a disease. Target identification draws on diverse data sources: genomic studies, proteomic analyses, clinical observations, and published literature linking molecular mechanisms to disease pathology.
Validation is the next critical step. Identifying a target is not the same as proving it is “druggable” or therapeutically relevant. Validation involves demonstrating, through experimental models, that modulating the target produces a measurable therapeutic effect. Without robust validation, downstream efforts risk being built on a flawed hypothesis.
Criteria for Target Selection: Disease Link, Predicted Safety, Biomarkers
Not all validated targets are equally suitable for drug development. Key selection criteria include:
- Strength of disease link: Is the target a driver of the disease or merely associated with it?
- Predicted safety: Targets expressed broadly across healthy tissues may produce unacceptable off-target effects.
- Biomarker availability: A target with no feasible biomarker readout makes clinical trial design significantly more challenging and expensive.
What Is Hit Discovery (Screening) and How Does It Work?
Once a target is validated, the search for molecules that interact with it begins. “Hit discovery” involves screening large libraries of chemical compounds or biological molecules to find “hits” — molecules showing preliminary activity against the validated target. The scale of this effort can be enormous. High-throughput screening (HTS) allows researchers to test hundreds of thousands of compounds in automated assay systems within days.
Alternative approaches include virtual screening, where computational models predict which molecules are likely to bind the target, and fragment-based screening, which identifies small molecular fragments that can be chemically elaborated into larger, more potent compounds. Regardless of the method, hits are starting points — not finished products. They require extensive verification, counter-screening against related targets, and confirmation of dose-response relationships before advancing further.
What Is Hit-to-Lead and What Is Its Goal?
The hit-to-lead phase transforms raw screening hits into “leads” — compounds with improved potency, selectivity, and early drug-like properties. This is where medicinal chemistry becomes central. Chemists systematically modify the chemical structures of hits, guided by structure-activity relationship (SAR) data, to enhance their interaction with the target while reducing interactions with unrelated biological systems.
The goal is not to produce a final drug candidate. Rather, it is to identify one or more chemical series with sufficient promise — in terms of activity, selectivity, and preliminary ADME characteristics — to justify the significant investment required for lead optimization. At this stage, early in vitro biological testing plays a decisive role. Da-Ta Biotech’s cell-based assay capabilities, for example, enable researchers to assess compound activity in biologically relevant tissue culture models, providing actionable data that guides SAR decisions efficiently.
From 50,000 Compounds to 3 Lead Series
An early-stage oncology biotech used Da-Ta Biotech’s validated cell-based assay platform to screen a targeted library of 50,000 compounds. Within 8 weeks, 47 hits were confirmed, refined to 6 chemical series, and ultimately narrowed to 3 leads with IC50 values below 10 nM and >100-fold selectivity over related kinases.
Early ADME Profiling Saves 18 Months
A CNS drug program integrated in vitro permeability and metabolic stability assays into the hit-to-lead phase, identifying a metabolic soft spot that would have been catastrophic in vivo. Early structural modification resolved the liability, preventing an estimated 18-month delay if discovered at the PK study stage.
Lead Optimization: Which Parameters Are Improved and How?
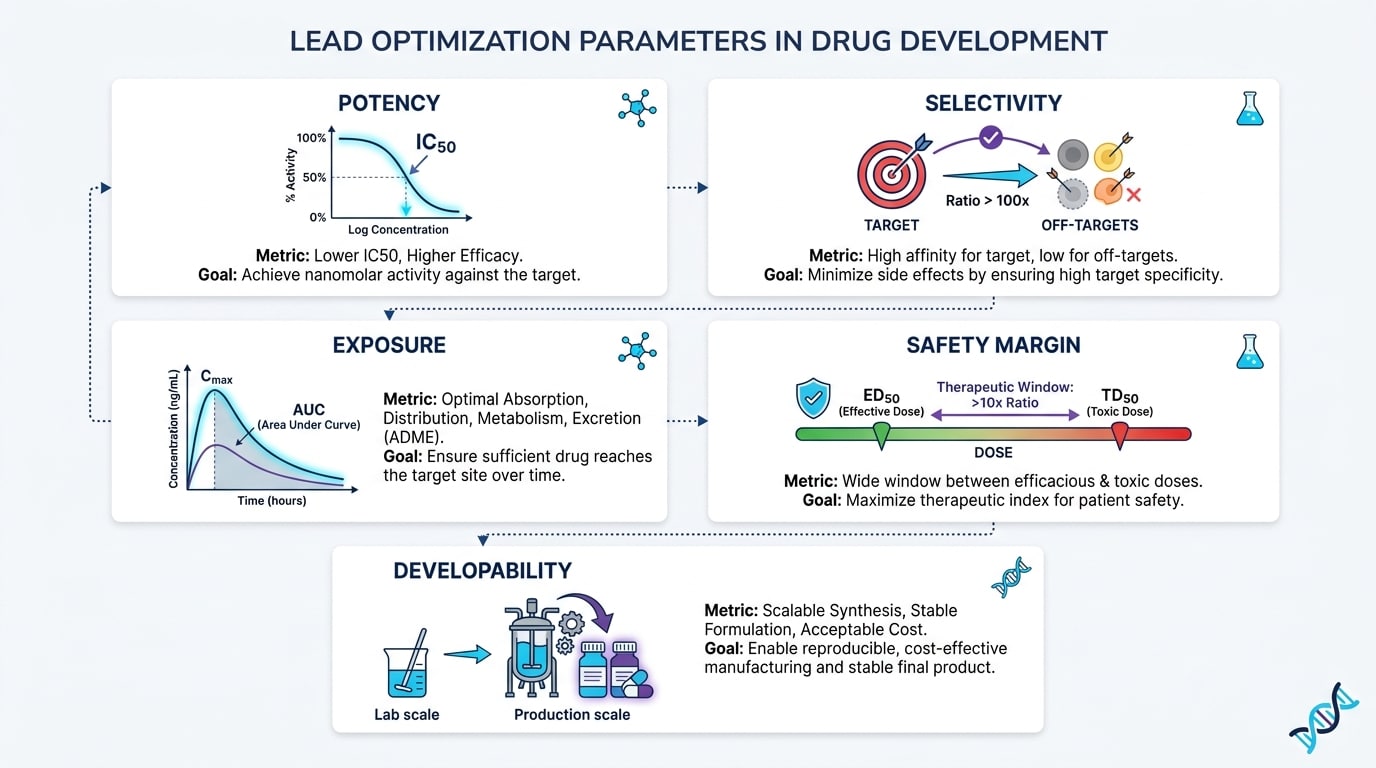
Lead optimization is an iterative, resource-intensive process where lead compounds are systematically refined. The objective is to produce a molecule — or a small number of molecules — with the best possible balance of efficacy, safety, and developability. Parameters that are actively improved during this phase include potency against the target, selectivity over related targets, and ADME (absorption, distribution, metabolism, excretion) properties that predict favorable pharmacokinetic behavior in humans.
Toxicity profiling begins here as well: early signals of genotoxicity, cytotoxicity, or organ-specific toxicity can eliminate a compound before larger investments are made. Developability — the practical feasibility of manufacturing the compound at scale, its chemical stability, and its formulation options — is assessed in parallel. The pharma development process demands that optimization decisions are not made in isolation; every change to improve one parameter must be evaluated for its impact on all others.
Key Metrics: Potency, Selectivity, Exposure, Safety Margin, Developability
When Is a “Candidate Selected” and What Does It Actually Mean?
Candidate selection is the single most consequential decision in early drug development. It is the point where one or a few lead compounds are formally chosen to advance into preclinical drug development — the IND-enabling phase. This decision is not made lightly. It is based on a comprehensive data package that includes potency and selectivity data, in vitro and in vivo ADME/PK profiles, preliminary toxicology results, and an initial assessment of manufacturability.
Programs that rush candidate selection, or make it based on incomplete data, frequently encounter costly failures in later drug development stages. Candidate selection represents a commitment of significant financial, operational, and scientific resources to a specific molecule. The data generated during discovery directly determines whether this decision is well-founded or premature.
What Happens in Preclinical Drug Development (IND-Enabling) Step-by-Step?
Preclinical drug development is the non-human testing phase designed to assess the safety and biological activity of the selected drug candidate in sufficient depth to justify first-in-human exposure. The primary goal is to generate a data package that supports an Investigational New Drug (IND) application. Activities during this phase include comprehensive pharmacokinetics (PK) studies, pharmacodynamics (PD) assessments, detailed ADME characterization, and formal toxicology programs.
The IND-enabling phase is also where CMC development intensifies — producing the drug substance and drug product under controlled conditions suitable for clinical use. Regulatory strategy crystallizes during this period as well, with teams determining the appropriate clinical starting dose, defining the first-in-human study design, and preparing the documentation that will be submitted to regulatory authorities. This is one of the drug development pipeline stages where experienced CRO partners provide the most value, offering validated assay platforms and robust protocols that generate regulatory-grade data.
Scientific Preclinical Research vs. Regulatory GLP Toxicology
Not all preclinical work carries the same regulatory weight. Early exploratory studies — screening assays, mechanism-of-action experiments, preliminary PK studies — are conducted under standard laboratory conditions and serve to inform scientific decisions. These studies are essential but are not typically submitted as pivotal data in an IND application.
Formal GLP (Good Laboratory Practice) toxicology studies, by contrast, must comply with strict regulatory standards governing study conduct, documentation, quality assurance, and data integrity. GLP studies are the ones regulators rely on to assess whether it is safe to expose humans to the compound. The distinction is critical: scientific preclinical research guides internal decisions, while GLP toxicology studies enable regulatory decisions. Both are addressed in the ICH M3(R2) guideline, which outlines which nonclinical safety studies are recommended to support each clinical development phase.
⚡ Common Mistakes That Derail Preclinical Programs
- Underestimating CMC timelines — Assuming manufacturing clinical-grade compound is a simple scale-up of laboratory synthesis.
- Delaying regulatory interaction — Waiting until IND-ready rather than engaging early for pre-IND guidance.
- Siloing in vitro and in vivo workstreams — Discrepancies between cell-based assay results and animal model outcomes need explanation before submission, not during regulatory review.
- Selecting CROs based solely on cost — Scientific capability and regulatory experience are non-negotiable when data integrity is at stake.
What Is Checked in ADME/PK/PD Before Entering the Clinic?
Before a drug candidate can be tested in humans, researchers must understand how the body handles the molecule and how the molecule affects the body. ADME studies answer the first question: Is the compound absorbed into the bloodstream? How is it distributed across tissues? Which enzymes metabolize it, and what metabolites are formed? How and how quickly is it excreted?
Pharmacokinetics (PK) quantifies these processes over time, determining peak drug concentrations, half-life, and total exposure. Pharmacodynamics (PD) examines the drug’s biochemical and physiological effects — its mechanism of action at the molecular level and the relationship between drug concentration and therapeutic response. Together, ADME, PK, and PD data predict appropriate human dosing, dosing frequency, and potential drug-drug interactions.
Da-Ta Biotech’s in vitro ADME and cell-based assay platforms provide researchers with critical ADME, PK, and PD data points early in the preclinical drug development process, enabling informed go/no-go decisions before committing to expensive in vivo studies. All assays are performed in an ISO 9001:2015-certified environment, ensuring data quality that meets regulatory expectations.
What Is GLP Toxicology and What Must It Prove?
GLP toxicology studies are the cornerstone of the safety assessment required before human exposure. These rigorously controlled studies must identify target organs for toxicity — which tissues or organ systems are most vulnerable to the compound’s effects. They must establish a No Observed Adverse Effect Level (NOAEL), which is used to calculate a safe starting dose for first-in-human trials.
GLP studies must also define the safety margin: the ratio between the dose that causes toxicity in animals and the expected therapeutic dose in humans. A narrow safety margin may make a candidate unsuitable for clinical development, regardless of its efficacy. As detailed in a comprehensive guide, GLP toxicology studies are a critical component of preclinical drug development, ensuring that potential toxicities are thoroughly investigated before human trials. Study types typically include:
- Single-dose and repeat-dose toxicity studies
- Genotoxicity assessments (Ames test, in vitro and in vivo clastogenicity)
- Safety pharmacology covering cardiovascular, respiratory, and CNS effects
- Reproductive and developmental toxicology (if applicable)
“GLP toxicology is not a formality — it is the scientific contract between the laboratory and the regulator. Every deviation in study conduct is a potential clinical hold.”
— Dr. Rinat Borenshtain-Koreh, PhD, DVM | Biotech & Biomed R&D Expert
What Is CMC and Why Does It Matter Already in Early Stages?
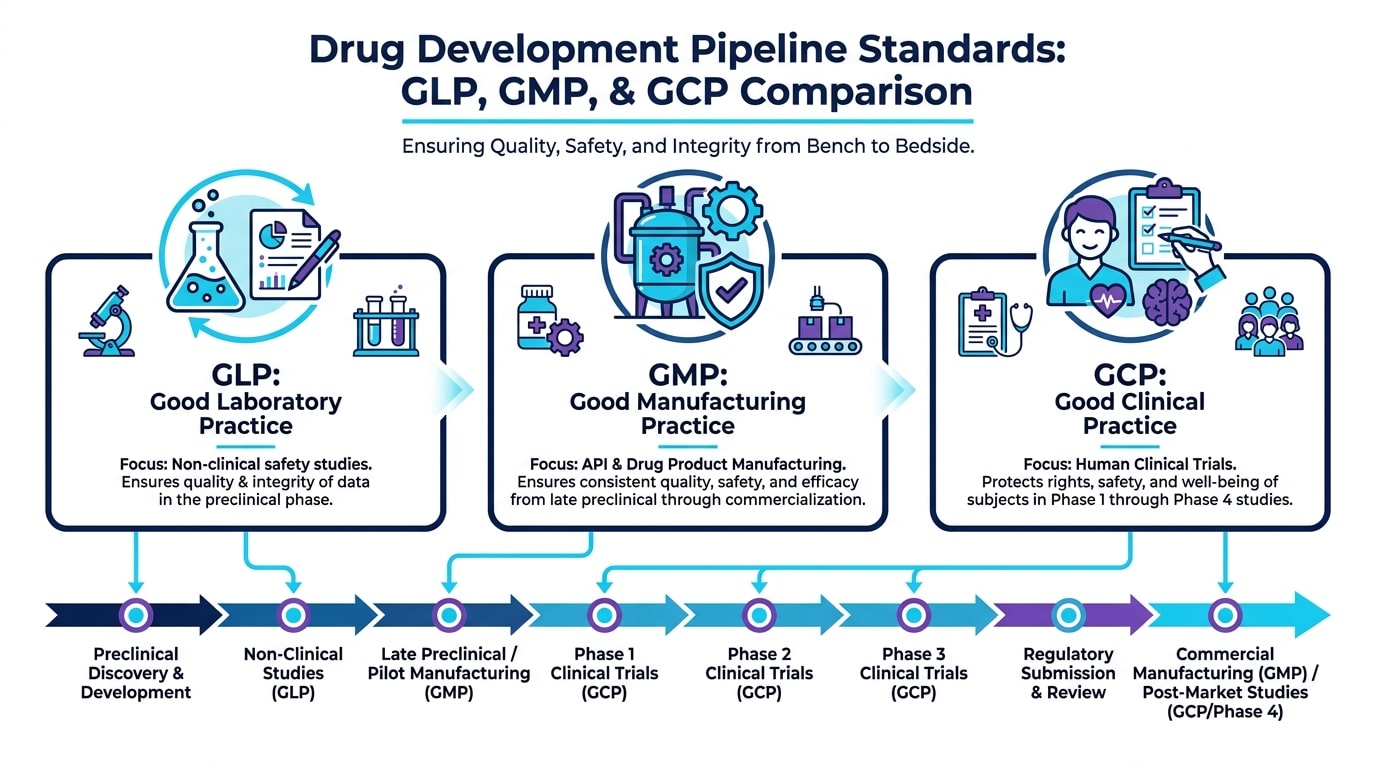
CMC (Chemistry, Manufacturing, and Controls) encompasses everything related to the chemical identity of the active pharmaceutical ingredient (API), the manufacturing process used to produce it, the quality control testing applied at each step, and the stability of both the API and the final drug product. Without robust CMC data, no IND can be filed and no clinical trial can begin.
CMC is often underestimated in early drug development stages because the focus tends to be on biological activity and safety. But manufacturing challenges discovered late — polymorphism issues, impurity profiles that change with scale, stability failures — can delay programs by months or even years. Starting CMC work early, in parallel with preclinical studies, ensures that clinical-grade material is available when needed and that the manufacturing process is sufficiently understood to satisfy regulatory requirements in the pharma development process.
The Difference Between GMP, GLP, and GCP
These three standards are complementary, not interchangeable. GLP ensures laboratory data integrity. GMP ensures manufacturing consistency and quality for investigational medicinal products used in clinical trials. GCP ensures ethical and scientific rigor in human studies. Understanding where each standard applies — and ensuring compliance at the right time — is fundamental to maintaining a viable regulatory pathway.
What Is an IND and When Is It Filed?
The Investigational New Drug (IND) application is a formal request to a regulatory authority — the FDA in the United States, the Israeli Ministry of Health in Israel, or equivalent bodies in other jurisdictions — for permission to begin human clinical trials. It is filed after sufficient preclinical drug development data has been generated to demonstrate that the drug candidate is reasonably safe for initial testing in humans and that the manufacturing process produces a consistent, well-characterized product.
An IND submission typically includes three major components:
Pharmacology, toxicology, and PK/ADME data demonstrating safety and biological activity in non-human models.
Drug substance and drug product characterization, manufacturing process description, quality controls, and stability data.
Proposed study protocols, investigator qualifications, and informed consent documents for the first-in-human trial.
The regulatory authority reviews the IND and, if no clinical hold is issued within 30 days (in the FDA pathway), the sponsor may proceed with clinical trials. More details on IND requirements are available directly from the FDA’s IND application page.
How Da-Ta Biotech Supports Critical Pipeline Needs
From early hit characterization to building investor-ready data packages, Da-Ta Biotech’s ISO 9001:2015-certified laboratory delivers validated, regulatory-grade data at every critical junction in the drug development pipeline.
ISO 9001:2015 Certified | Validated Protocols | Regulatory-Grade Output
Every assay performed at Da-Ta Biotech follows a validated, documented protocol with defined acceptance criteria. Our quality management system ensures full traceability from sample receipt through data delivery. Assay platforms are routinely qualified using reference compounds with established literature values, ensuring the data we deliver is reproducible, defensible, and suitable for regulatory submission packages.
Our team collaborates directly with client research teams to design studies that answer the right scientific questions at the right time — maximizing the value of every experimental investment in your pipeline.
Frequently Asked Questions
Ready to Move Your Pipeline Forward?
Are you developing a molecule and need reliable in vitro biological data to support your next gate decision — whether it is hit confirmation, lead optimization, ADME profiling, or building a data package for investors and regulatory submissions? Da-Ta Biotech’s experienced R&D team provides validated assay platforms and robust protocols designed to deliver the answers you need.

