
Biocompatibility Testing – ISO 10993
Dr. Rinat Borenshtain-Koreh, PhD & DVM — With over 25 years directing biocompatibility evaluations and biological safety assessments for medical devices, I’ve guided hundreds of companies through ISO 10993 compliance. As CEO of Da-Ta Biotech and Scientific Manager of Biotech Farm, I’ve seen what separates successful regulatory submissions from costly failures. Here’s the definitive guide to biocompatibility testing in Israel.
Expert Insight: The most expensive biocompatibility testing mistake isn’t a failed test — it’s submitting samples that don’t represent your finished device. I’ve seen projects delayed by months because companies tested raw materials instead of the sterilized, packaged product. Get the documentation right before any sample leaves your facility.
What is Biocompatibility Testing and Why is it Mandatory for Medical Devices?
Biocompatibility testing is the systematic evaluation of how a medical device interacts with biological systems — human tissues, cells, and body fluids. The goal is straightforward: determine whether the device can perform its intended function without causing unacceptable harm to the patient. This is not a theoretical exercise. It evaluates the finished product in its final form, accounting for manufacturing residues, sterilization processes, coatings, adhesives, and packaging materials.
Regulatory bodies worldwide mandate biocompatibility testing for any medical device that comes into direct or indirect contact with the body. The FDA requires a thorough biocompatibility assessment as part of premarket submissions. The European Medical Device Regulation (EU MDR) imposes comparable requirements. Without documented evidence of biological safety, a device simply cannot reach the market — regardless of how innovative or effective it may be.
Why Search for “Biocompatibility Testing Israel” — the Advantages of Local Testing
When Israeli medical device companies search for biocompatibility testing Israel, the intent is practical. International shipping of biological samples introduces delays, customs complications, and cold-chain risks. Working with a local partner eliminates most of these variables.
Choosing an Israeli-based laboratory like Da-Ta Biotech means reduced turnaround on communication — questions about sample preparation or protocol adjustments are resolved within the same business day and time zone. Sample logistics become simpler: packaging, temperature control, and transit times are minimized when the lab is within driving distance. Perhaps most importantly, a local partner understands the regulatory landscape Israeli companies navigate — whether targeting US FDA clearance, European CE marking, or both simultaneously.
Da-Ta Biotech’s facility in Rehovot’s Science Park provides experienced R&D experts who can sit with your team face-to-face during the critical planning stages, reducing costly misunderstandings that frequently occur when project details are communicated across continents.
How Does ISO 10993-1 Determine Which Biocompatibility Tests are Needed for My Device?
ISO 10993-1 is the foundational standard governing the biological evaluation of medical devices. It does not prescribe a rigid checklist. Instead, it defines a risk-based framework that categorizes your device according to two primary variables: the nature of body contact and the duration of that contact. These two variables together determine which biological endpoints must be evaluated.
The standard also emphasizes that testing should be the last resort, not the first step. Before committing to laboratory work, manufacturers are expected to review existing data — literature, material safety data sheets, chemical characterization results, and predicate device information. Only gaps remaining after this review should be addressed through physical testing. This approach, reinforced in the FDA’s guidance on ISO 10993-1, reduces unnecessary testing while ensuring comprehensive safety evaluation.
What Constitutes “Nature of Contact” — Intact Skin, Mucosa, Blood, Implant?
ISO 10993-1 classifies devices into categories based on where they contact the body. A wound dressing touching intact skin faces different biological risks than a catheter contacting mucous membranes. A vascular stent in direct blood contact triggers an entirely different set of endpoint requirements than an external monitoring electrode. Implantable devices — those placed within bone, tissue, or the cardiovascular system — demand the most extensive evaluation because they interact deeply and continuously with living systems.
What Constitutes “Duration of Contact” — Limited, Prolonged, Long-Term?
Duration categories are defined as follows: limited contact lasts less than 24 hours; prolonged contact spans 24 hours to 30 days; long-term or permanent contact exceeds 30 days. As duration increases, the required scope of biological evaluation escalates significantly. A device used for a few minutes during a procedure may only require cytotoxicity and irritation assessment, while a permanent implant demands systemic toxicity, genotoxicity, implantation studies, and more. The FDA’s endpoint tables organized by contact duration provide a practical reference for this escalation.
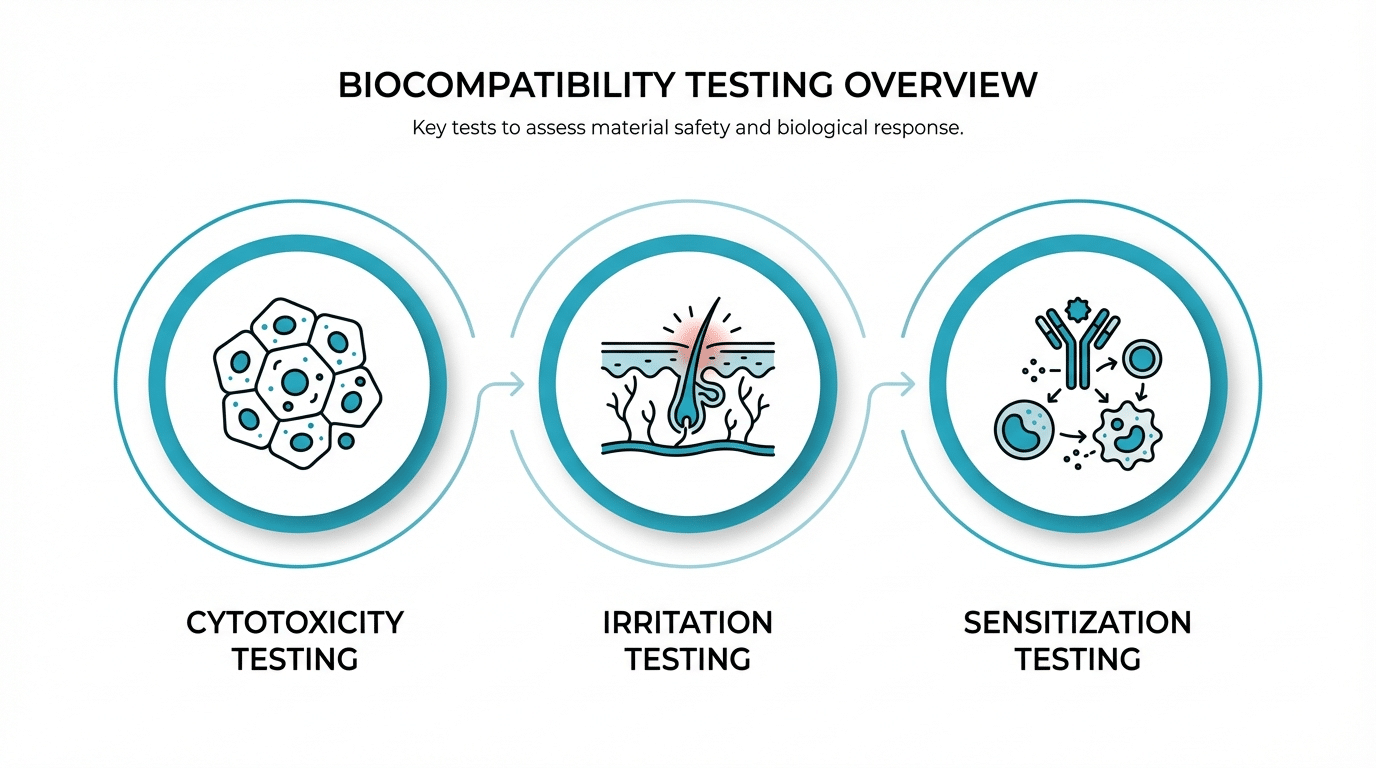
Which Tests Form the Baseline in Most Projects?
For nearly every medical device that contacts the body, three biological endpoints form the starting point: cytotoxicity, irritation, and sensitization. These are sometimes referred to as the “Big Three” of biocompatibility testing. They screen for the most fundamental biological risks — whether device materials kill cells, cause local inflammation, or trigger allergic reactions.
The exact selection and sequence still depend on your device’s ISO 10993-1 classification. A device touching only intact skin for limited duration may require only these three. A blood-contacting implant will require them plus many additional endpoints. Da-Ta Biotech specializes in a wide range of biological evaluations and cell-based assays for the biotechnology industry, providing the scientific infrastructure to execute these baseline tests with validated protocols under ISO 9001:2015 quality management.
The “Big Three” Baseline Tests
- Cytotoxicity (ISO 10993-5): In vitro cell viability assessment
- Irritation (ISO 10993-23): Local tissue reaction evaluation
- Sensitization (ISO 10993-10): Allergic response potential
What is Cytotoxicity Testing According to ISO 10993-5 and When Does it Fail?
Cytotoxicity testing under ISO 10993-5 is an in vitro assay that determines whether materials extracted from a device are toxic to living cells. Cultured mammalian cells are exposed to device extracts or placed in direct contact with the material. Cell viability is then measured — typically using quantitative methods such as MTT or XTT assays that assess metabolic activity.
This test is among the most sensitive screening tools available. It is required for virtually all devices with body contact and is often the first biological test performed. Common reasons for failure include residual manufacturing chemicals (mold release agents, machining oils), inadequate cleaning validation, aggressive sterilization residues (such as ethylene oxide), or inherently cytotoxic material additives. A failed cytotoxicity test is not merely a test failure — it signals a material or process problem that must be investigated and resolved before proceeding with additional endpoints.
⚠️ Warning: Cytotoxicity failures often trace back to insufficient ethylene oxide aeration after sterilization. If your device uses EtO sterilization, verify aeration parameters meet ISO 10993-7 residual limits before submitting samples for testing.
Irritation Testing Under ISO 10993-23 — Choosing the Right Model

Irritation testing evaluates whether a device causes localized inflammatory reactions at the contact site — skin redness, swelling, or tissue damage. ISO 10993-23 provides the framework for selecting appropriate test models. The choice depends heavily on the intended clinical contact site: skin irritation models for dermal devices, ocular models for ophthalmic devices, and mucosal models for devices contacting internal membranes.
Modern practice increasingly favors validated in vitro tissue models (reconstructed human epidermis, for example) over traditional animal-based methods. However, the test must accurately replicate the device’s real-world exposure conditions. Extract preparation — the solvents used, temperature, and duration — must mirror clinical use. Misaligned extraction conditions are a frequent source of irrelevant or misleading results.
Sensitization Testing — A Scenario Where Delays Frequently Occur
Consider this scenario: a medical device company completes cytotoxicity and irritation testing successfully, then submits samples for sensitization testing — only to discover the finished device contains a new adhesive component introduced during a recent design change. The test fails. Weeks of work must be repeated.
Sensitization testing assesses a device’s potential to provoke allergic or hypersensitivity reactions after repeated or prolonged exposure. Device failures in this test frequently trace back to material additives (plasticizers, colorants, antioxidants), process residues, or changes in supplier formulations that were not communicated to the testing lab. Clear documentation of every component in the finished device — and immediate notification of any design or process changes — is essential. The testing lab needs to evaluate exactly what the patient will encounter, not an approximation.
When is Hemocompatibility Testing Critical?
Hemocompatibility testing under ISO 10993-4 evaluates how a device interacts with blood. It is required for any device with direct or indirect blood contact — catheters, infusion sets, dialysis components, blood collection systems, vascular grafts, heart valves, and extracorporeal circulation equipment. The evaluated endpoints can include hemolysis (destruction of red blood cells), thrombosis (blood clot formation), coagulation pathway activation, platelet adhesion, and complement system activation.
The scope of hemocompatibility testing can be extensive. A simple blood collection needle requires less evaluation than a permanent cardiovascular implant. The test matrix, sample preparation, and blood handling conditions must be carefully designed — fresh human blood is typically required, and the logistics of procurement and testing within defined time windows add complexity to the process.
Chemical Characterization and Extractables & Leachables — Why Regulators Increasingly Demand Them
Chemical characterization and Extractables & Leachables (E&L) studies identify and quantify substances that can migrate from a medical device into the body or surrounding environment. Extractables are compounds released under exaggerated laboratory conditions; leachables are those released under conditions simulating actual clinical use.
This data has become increasingly central to biocompatibility evaluation. The FDA’s evolving guidance places growing emphasis on chemical analysis as a complement — and sometimes an alternative — to biological testing. When E&L data is combined with toxicological risk assessment (per ISO 10993-17), manufacturers can build a comprehensive safety argument that may reduce the need for certain animal studies. E&L studies also serve a diagnostic function: if a device fails a biological test, chemical characterization can help identify the specific substance responsible.
How Long Does Biocompatibility Testing Actually Take?
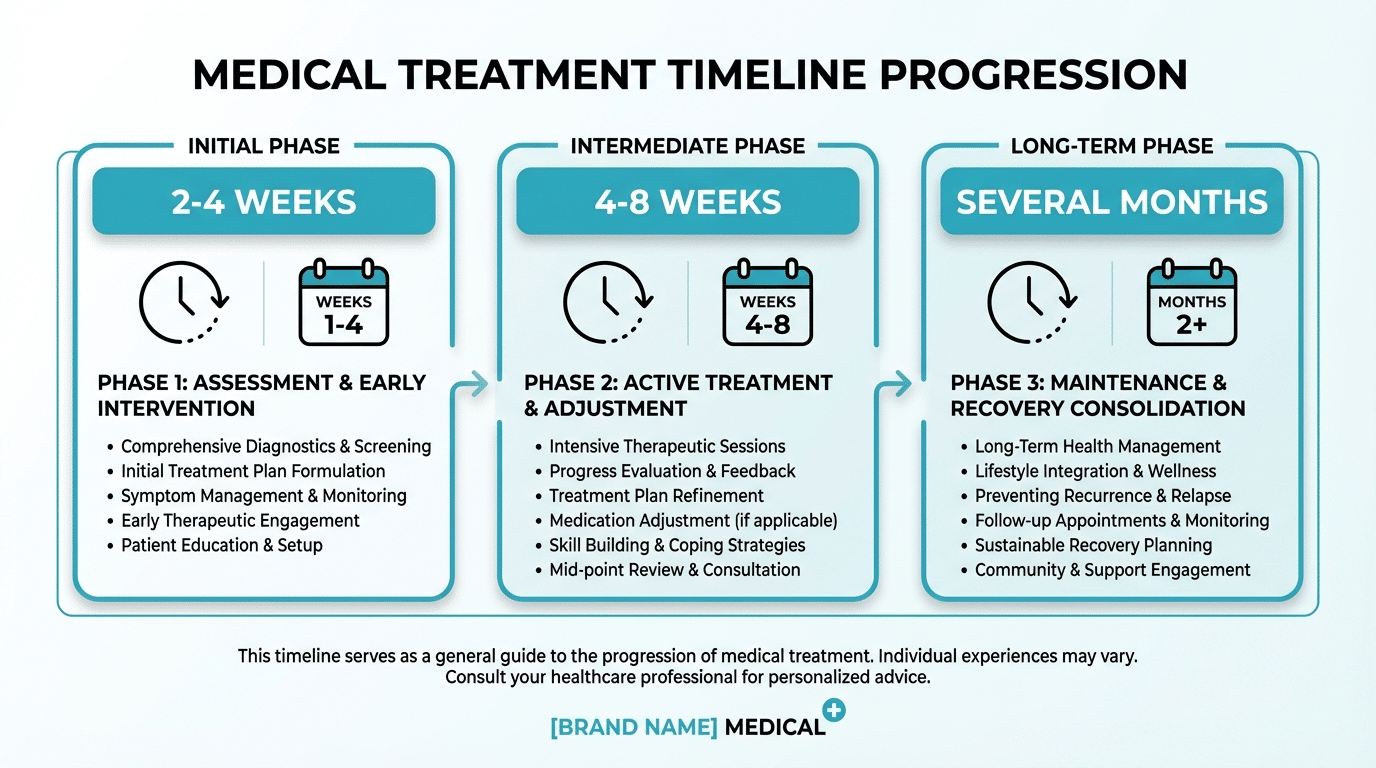
Timelines vary dramatically. A straightforward in vitro cytotoxicity test can be completed within 2–4 weeks from sample receipt. Irritation and sensitization testing may take 4–8 weeks. Hemocompatibility, systemic toxicity, and implantation studies can extend to several months. Chemical characterization timelines depend on the complexity of the device and the number of extraction conditions required.
The most common cause of timeline overruns is not the testing itself — it is inadequate preparation. Samples that do not represent the finished device, missing documentation, incomplete material declarations, and mid-project design changes all force retesting. “Second rounds” of testing are expensive and time-consuming. Investing time in thorough pre-test planning consistently saves months on the back end. Da-Ta Biotech’s approach to pre-test consultation — reviewing device specifications, sample adequacy, and regulatory requirements before any lab work begins — is specifically designed to prevent these delays.
Typical Testing Timelines
- Cytotoxicity (in vitro): 2–4 weeks
- Irritation testing: 4–6 weeks
- Sensitization testing: 4–8 weeks
- Hemocompatibility: 6–12 weeks
- Chemical characterization: 4–10 weeks (complexity dependent)
Cost Factors for Biocompatibility Testing in Israel
Biocompatibility testing costs are driven by the scope of the biological evaluation, not by a fixed price list. The primary cost determinants include the number and type of biological endpoints required, the complexity of sample extraction and preparation, whether GLP (Good Laboratory Practice) compliance is mandated, the number of device variants or configurations to be tested, and the depth of reporting needed for the specific regulatory submission.
In vitro tests are generally less expensive than in vivo studies. A device requiring only the three baseline tests (cytotoxicity, irritation, sensitization) will cost substantially less than an implantable device requiring systemic toxicity, genotoxicity, implantation, and hemocompatibility evaluations. Working with a partner who provides clear cost breakdowns during the planning phase — and who identifies unnecessary tests before they are performed — protects your budget without compromising the regulatory acceptability of your data.
What Documents Should You Prepare Before Starting — the Mistake That Costs the Most Time
The single most common mistake in biocompatibility testing is submitting samples without adequate documentation. The laboratory needs comprehensive information to design the correct test protocol, and regulators will scrutinize whether the tested article truly represents the marketed device.
You should prepare the following before contacting the lab: a detailed description of the device’s intended use and clinical application; a complete Bill of Materials (BOM) listing every component, adhesive, coating, ink, and packaging material; a description of all manufacturing processes including cleaning and sterilization; and clear identification of the final, finished configuration that will be tested. Inconsistencies between what is documented and what is physically submitted are a leading cause of regulatory rejection.
Biocompatibility Testing Versus Biological Evaluation — a Critical Distinction
These terms are frequently used interchangeably, but they refer to different things. Biocompatibility testing means the physical laboratory tests performed on a device — cytotoxicity assays, irritation studies, sensitization evaluations. Biological evaluation is the entire process defined by ISO 10993-1, which encompasses risk assessment, literature review, chemical characterization, the tests themselves, toxicological risk assessment, and the final documented conclusion.
Testing is a component of biological evaluation, not a substitute for it. A manufacturer who performs all required tests but fails to document the rationale, review existing data, or compile a coherent evaluation report will face regulatory pushback. Understanding this distinction from the outset saves significant rework during submission preparation.
Do You Need a Biological Evaluation Plan (BEP)?
Yes. A Biological Evaluation Plan (BEP) is a documented strategy that outlines the entire approach to evaluating your device’s biological safety. Both the FDA and EU Notified Bodies expect to see a BEP as part of the regulatory submission. The BEP specifies the device description, material composition, body contact classification, applicable ISO 10993 parts, selected evaluation endpoints, and — critically — the rationale for including or excluding specific tests.
A well-constructed BEP prevents two common problems: performing tests that are not required (wasting time and budget) and omitting tests that are required (triggering regulatory deficiency letters). The BEP should be developed collaboratively between the device manufacturer and the testing laboratory, ideally before any samples are submitted.
How is a Biological Evaluation Report (BER) Structured?
The Biological Evaluation Report (BER) is the final document that synthesizes all elements of the biological evaluation into a single, coherent narrative. It is this document — not individual test reports — that regulators review to assess whether a device is biologically safe.
A typical BER includes an executive summary, a complete device description with intended use and materials, the results of chemical characterization and literature reviews, detailed summaries of all performed biological tests, a toxicological risk assessment for any identified leachables, and an overall conclusion on the device’s biological safety profile. The BER must clearly trace the logic from the BEP through the data to the conclusion. Gaps, inconsistencies, or missing justifications are common grounds for regulatory queries during 510(k) or CE marking reviews.
Can Biocompatibility be Demonstrated Without Animal Testing?
Increasingly, yes — but not universally. Modern regulatory philosophy, embedded in both ISO 10993-1 and FDA guidance, explicitly encourages the use of non-animal methods wherever scientifically justified. In vitro cytotoxicity assays, reconstructed tissue models for irritation, chemical characterization combined with toxicological risk assessment, and thorough literature reviews can collectively address many biological endpoints without animal involvement.
However, certain complex endpoints — particularly chronic systemic toxicity, some aspects of hemocompatibility, and long-term implantation responses — may still require in vivo studies when no validated alternative exists. The key is building a data package that is comprehensive enough to justify each decision. Da-Ta Biotech is committed to ethical and efficient testing practices, providing robust in-vitro testing solutions that align with the global regulatory shift toward reducing animal use while maintaining the scientific rigor regulators expect.
✓ Regulatory Trend: Both FDA and EU MDR increasingly favor in vitro methods and chemical characterization over animal testing when scientifically justified. A well-documented rationale in your BEP supporting non-animal approaches is now viewed positively by regulatory reviewers.
Five Common Pitfalls in Biocompatibility Testing — and What Goes Wrong
Testing unrepresentative samples tops the list. Companies sometimes submit raw material coupons or prototype components instead of the finished, sterilized, packaged device. Regulators expect data on the article as it reaches the patient. Inadequate BEP documentation is the second pitfall — a vague or missing plan leads to redundant testing or critical gaps discovered only during regulatory review.
Ignoring process impacts ranks third. A biocompatible polymer can become cytotoxic after ethylene oxide sterilization if aeration is insufficient. Fourth, undocumented supplier or design changes midway through a project invalidate prior test results. Fifth, misinterpreting endpoint requirements — selecting tests based on a generic table rather than a device-specific risk assessment — creates data packages that do not satisfy regulatory scrutiny.
Da-Ta Biotech’s pre-test consultation process directly addresses each of these pitfalls. Experienced R&D experts review your device configuration, documentation, and regulatory target before proposing a testing strategy. This proactive approach identifies potential issues early — when they are inexpensive to correct — rather than after testing has begun. For insights into comprehensive R&D services for the biotech and biomed industries, and to understand our full capabilities, reach out to our team.
Pitfall #1: Unrepresentative Samples
Submitting raw materials or prototypes instead of final, sterilized, packaged devices. Test what the patient will encounter.
Pitfall #2: Inadequate BEP
A vague or missing Biological Evaluation Plan leads to redundant testing or gaps discovered during regulatory review.
Pitfall #3: Ignoring Process Impacts
Sterilization, cleaning, and manufacturing residues can transform biocompatible materials into cytotoxic ones.
Mapping Your Needs to Da-Ta Biotech’s Capabilities
Frequently Asked Questions About Biocompatibility Testing
Have a Scientific Challenge? Da-Ta Biotech Provides the Tools to Address It
Whether you are planning your first biocompatibility evaluation or troubleshooting a failed test result, the path forward starts with a clear conversation about your device, your regulatory targets, and your timeline. Da-Ta Biotech’s experienced R&D team works as your scientific partner — from proof-of-concept through regulatory submission — providing validated in-vitro testing, pre-test consultation, and the biological expertise needed to navigate ISO 10993 with confidence. Learn more about our biological laboratory for R&D in Israel and how we support biotech and medical device companies at every stage.
“Come to us with any scientific challenge — we are here for you. Our team doesn’t just run tests; we partner with you to solve complex biocompatibility questions and get your device to market.”
— Da-Ta Biotech Team
Ready to scope your project? Contact Da-Ta Biotech at in-vitro@databiotech.co.il or call +972-8-9100575.

