
Protein Binding Assay Services: A Practical Guide for Drug Developers
Reviewed & Written by Dr. Rinat Borenshtain-Koreh, PhD, DVM
With over 25 years of hands-on biotech and biomed R&D experience — spanning biological model development, in-vitro assays, in-vivo experiments, and FDA application support — Dr. Borenshtain-Koreh leads the team at Da-Ta Biotech LTD. The assertion is direct: a molecule whose binding profile is incompletely characterized will fail in vivo, not because it cannot bind, but because no one measured it properly.
⚗️ Expert Insight: Why Binding Data Can Make or Break Your IND
Free drug theory is not a theoretical abstraction — it is the physiological gatekeeper of your program. Only the unbound fraction (fu) crosses membranes, engages targets, and drives clearance. Without a complete binding profile covering both target engagement and plasma sequestration, exposure-response models are built on assumptions. The single most common preventable cause of late-stage IND surprises is binding data that was collected too late, too narrowly, or under unvalidated conditions.
What Do Professional Protein Binding Assay Services Include?
Lead optimization rarely fails because a molecule cannot bind — it fails because the binding profile was never fully characterized. Professional protein binding assay services close that gap by quantifying both equilibrium affinity (KD) and physiological kinetics, so program teams can prioritize candidates with realistic in vivo behavior. At Da-Ta Biotech, the workflow spans feasibility studies, custom assay development, parameter tuning, execution, and bioanalytical reporting — all under one validated roof in Rehovot Science Park.
Two pipelines run in parallel: one for target engagement verification (does your molecule bind the intended receptor or enzyme?) and one for matrix-binding profiles (how does plasma sequester it?). Combining both gives a defensible package for IND-enabling work — and that is the correct thing to do.
What Is a Protein Binding Assay?
A protein binding assay is an in vitro analytical technique that measures the physical association between a ligand — a small molecule, peptide, or antibody — and a protein. Think of it as a key-and-lock test, but with thousands of keys diffusing in solution, some sliding into the lock, others slipping away. The assay’s job is to count which keys are docked and which are free.
Critically, the measurement separates free (unbound) ligand from bound complexes. From that separation, scientists calculate fractional values (fu, percent bound) or kinetic constants. The lesson for non-experts: total drug concentration in a vial does not equal active pharmacological exposure — only the unbound portion engages the target.
How Do You Measure Drug–Protein Binding in Practice?
Measuring binding splits into two structurally different experimental designs. Target-binding assays assess affinity directly at the pharmacological site of action — receptors, kinases, ion channels, antibody epitopes. Matrix-binding assays evaluate non-specific integration with circulating transport elements, primarily Human Serum Albumin (HSA) and Alpha-1-Acid Glycoprotein (AGP).
Equipment diverges accordingly. Target-binding leans on real-time biosensing (SPR, BLI) where association and dissociation are tracked live. Matrix-binding leans on physical separation (dialysis, ultrafiltration) followed by analytical chromatography (LC-MS/MS). Both approaches feed into a complete protein-drug interaction profile, mapping how a compound performs from receptor docking to systemic clearance.
Why Plasma Protein Binding Matters for Drug Development
Free drug theory states a simple physiological truth: only the unbound fraction (fu) can cross cellular membranes, engage targets, and undergo metabolic clearance. The bound fraction is essentially parked on albumin or AGP, pharmacologically silent until released.
That matters because high plasma binding reshapes volume of distribution (Vd), half-life (t1/2), and toxicity margins. Consider a compound that is 99% bound versus one that is 98% bound — a difference that looks trivial on paper. In circulation, the active free concentration doubles. For highly bound drugs, that single percentage point can flip a safe exposure window into a toxic one, which is precisely why drug binding studies belong in IND-enabling packages, not late-stage afterthoughts.
“Only the unbound fraction engages the target, drives clearance, and determines toxicity. Measuring total drug concentration without knowing fu is like measuring fuel in a tank without knowing if the engine is connected.”
— Dr. Rinat Borenshtain-Koreh, PhD, DVM | Da-Ta Biotech LTD
Which Method Should I Choose: Equilibrium Dialysis vs. Ultrafiltration vs. Ultracentrifugation?
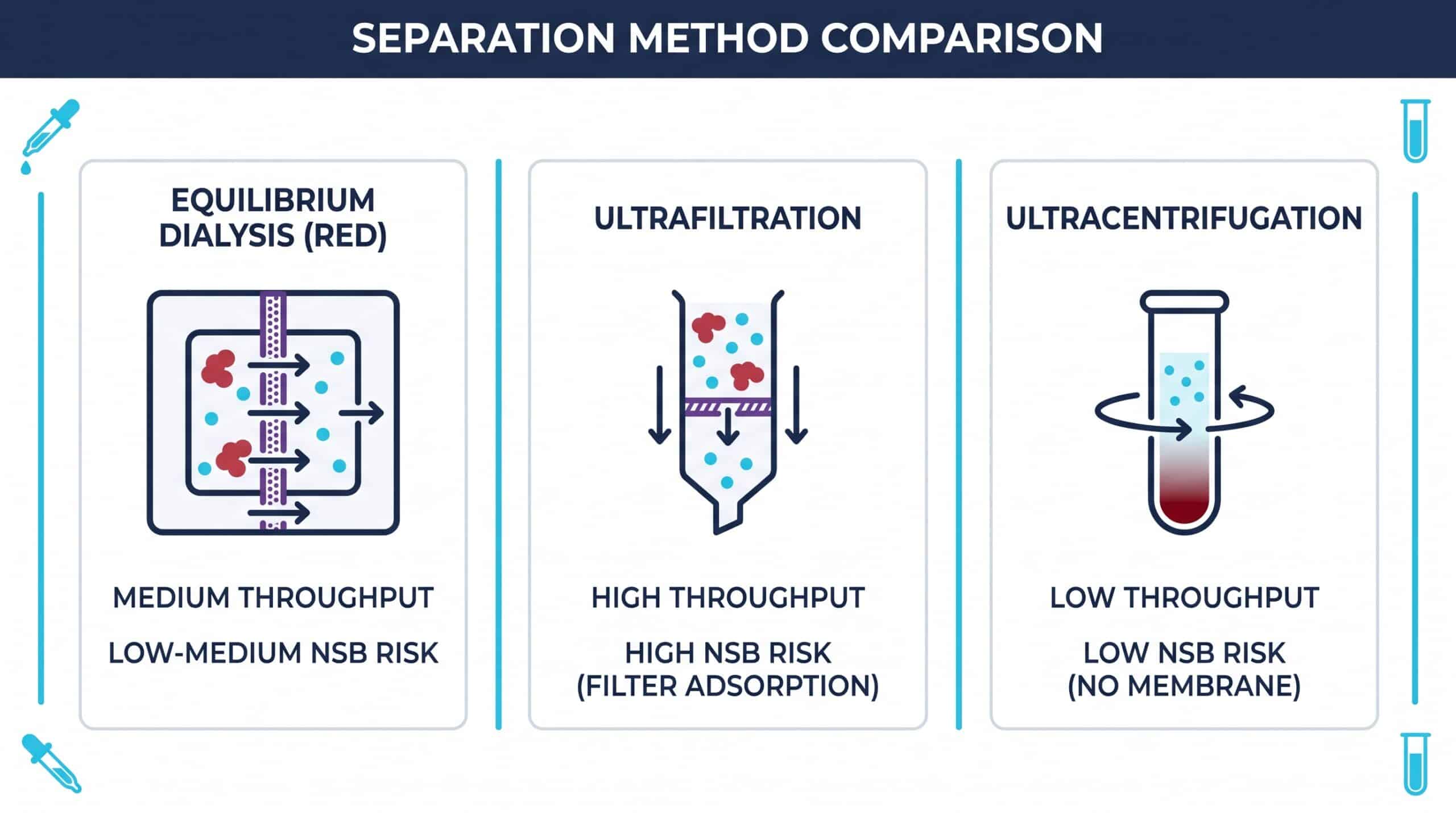
Selecting the right separation method depends on molecule stability, expected affinity range, lipophilicity, and assay throughput needs. No single technique is universally superior — each carries trade-offs that influence fu accuracy.
Equilibrium Dialysis (The Gold Standard)
Modern Rapid Equilibrium Dialysis (RED) devices use a porous semi-permeable membrane that segregates plasma from buffer. Unbound molecules diffuse freely until concentrations equilibrate. RED is widely regarded as the reference method because it avoids volume shifts that distort binding equilibria.
Ultrafiltration (The Speed-Focused Choice)
Centrifugal force pushes unbound molecules through a molecular-weight cutoff filter, leaving bound complexes behind. It is fast — minutes rather than hours — but adsorption of lipophilic compounds onto the filter membrane is a well-documented limitation that can mask true fu. Pre-saturation strategies (filter membrane adsorption review) mitigate the issue.
Ultracentrifugation (The Membrane-Free Alternative)
High-velocity centrifugal forces sediment macromolecules at the tube base without any membrane. This eliminates filter-adsorption artifacts entirely, though it requires specialized rotors and longer run times.
Affinity (KD) vs. Kinetics (kon/koff): What Is the Difference?
Affinity and kinetics describe the same interaction from two angles. The Equilibrium Dissociation Constant (KD) is a static ratio — it tells you where the binding sits at equilibrium. The kinetic constants kon (association rate) and koff (dissociation rate) describe the dynamic pathway: how quickly the complex forms and falls apart.
Why does this distinction matter in protein drug interaction work? Because two compounds with identical KD can behave very differently in vivo. Target residence time (1/koff) often predicts efficacy duration better than KD alone — a slow koff means the drug stays engaged with its target long after free plasma levels have dropped. Moderate-affinity compounds with slow dissociation can outperform high-affinity, fast-off competitors.
KD (Equilibrium Affinity)
Static snapshot of binding strength at equilibrium. Lower KD = higher affinity. Used to rank and compare candidates for intrinsic potency in early-stage prioritization.
kon (Association Rate)
How fast the drug-target complex forms. Diffusion-limited ceiling ~106–107 M-1s-1. Important for fast-acting mechanism requirements and onset of action predictions.
koff & Residence Time
How slowly the complex dissociates (1/koff = residence time). Slow koff extends pharmacological effect duration beyond plasma half-life — often the most actionable differentiator in lead selection.
Biosensor Assays for Small Molecules: SPR and BLI
When the analyte is a 300-Da small molecule and the target is a 60-kDa protein, signal generation becomes the central design problem. The mass difference means the binding event produces only a small detectable change, so sensor sensitivity and immobilization density must be carefully optimized.
Surface Plasmon Resonance (SPR)
SPR detects refractive index changes on gold-plated sensor chips as molecules bind to immobilized targets. The technique is label-free and operates in real time, producing sensorgrams that yield KD, kon, and koff in a single experiment. A recent SPR review details signal tracing advances and current sensitivity limits.
Biolayer Interferometry (BLI)
BLI uses fiber-optic sensors in a “dip-and-read” format, tracking changes in interference patterns as a biological layer thickens on the tip. It is well suited for crude samples and parallel screening, though small-molecule analytes typically require higher immobilization densities to lift signal above baseline noise.
🔬 SPR vs. BLI: Practical Selection Guidance
- SPR: Higher sensitivity for small molecules; lower baseline noise; preferred for accurate KD and kinetic constant determination.
- BLI: Higher tolerance for crude or complex samples; better for high-throughput parallel screening; requires higher target loading densities for small molecules.
What Sample Amount and Concentration Do You Need?
A complete binding study requires serial dilutions — typically 5 to 8 concentration points — to map a full binding curve. Skipping points compromises curve fitting; saturating too early misses the high-affinity regime.
Before submitting samples, document: compound purity (≥95% recommended), exact molecular weight, solubility in aqueous buffer, vehicle tolerance (DMSO usually capped at 1%), and any known stability concerns. Missing this information is the leading cause of sample precipitation and false-negative signals during execution. Treat the intake form as a scientific contract, not paperwork.
⚠️ Sample Submission Checklist — Do Not Skip Any Item
- Compound purity ≥ 95% (HPLC or NMR verified)
- Exact molecular weight and structure (SDF or SMILES preferred)
- Solubility in PBS/buffer and DMSO tolerance ≤1%
- Known stability concerns (light sensitivity, hydrolysis, oxidation)
- Expected KD range (if known from prior assays or in silico modeling)
What Deliverables Will I Receive from Drug Binding Studies?
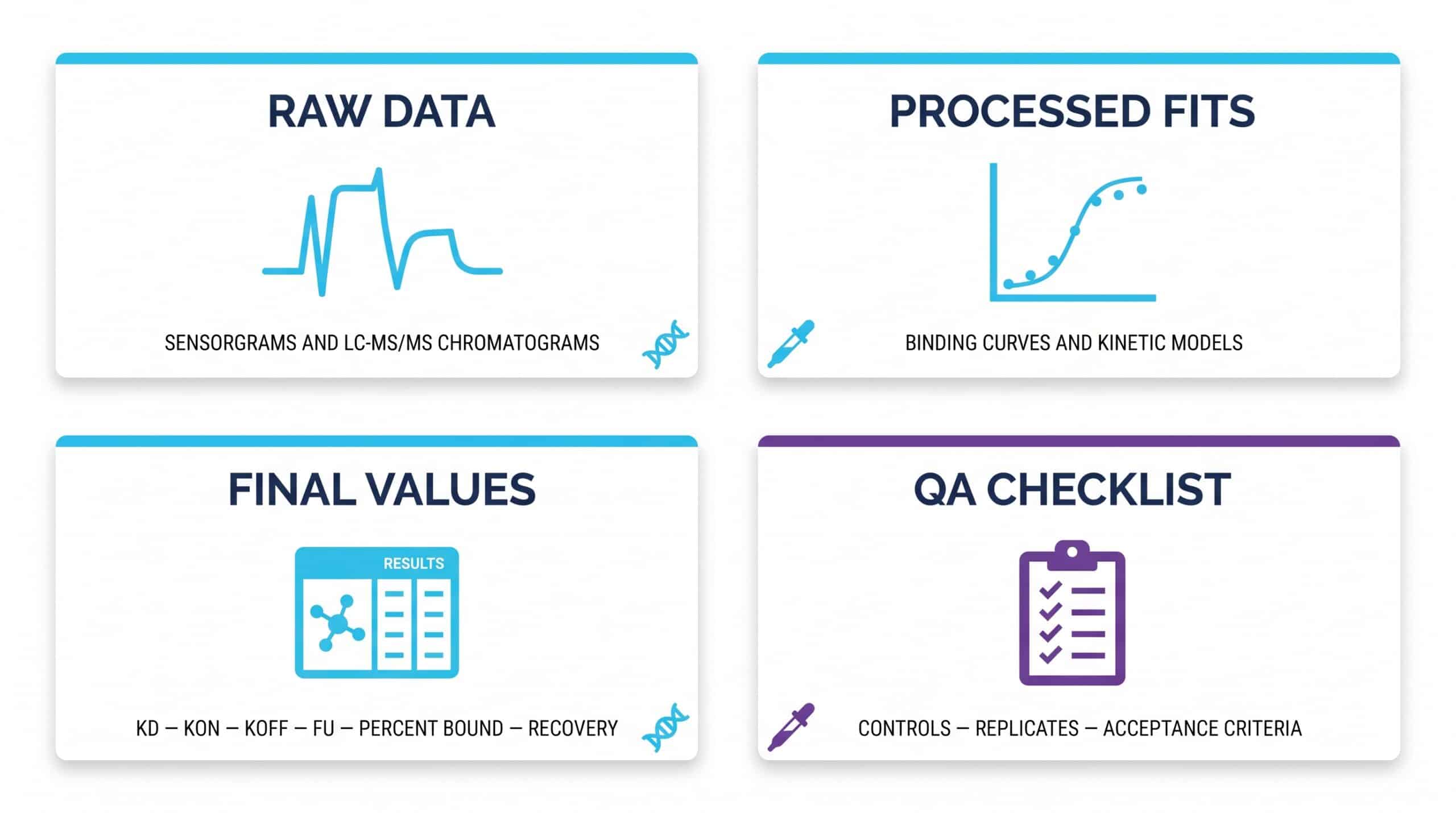
Professional drug binding studies deliver a layered report, not just a single number. Expect raw biosensor traces or chromatographic area measurements, processed curve fits, calculated final values, and the QA documentation that proves run validity.
Da-Ta Biotech Advantages Mapped to Common R&D Needs
How Do You Ensure Data Quality (Controls, Replicates, Validation)?
Data quality is built, not assumed. Every robust protocol includes positive reference control compounds with established binding benchmarks — typically warfarin or propranolol for plasma binding work — to confirm the system performs within expected ranges. Replicates (minimum n=3) detect outliers and quantify precision.
Active recovery verification confirms the compound is not lost to adsorption or degradation during the run. Validation parameters follow industry-standard bioanalytical frameworks aligned with the FDA M10 guideline: precision, accuracy, selectivity, and baseline tracking. These elements are non-negotiable for regulatory submissions.
🧪 Da-Ta Biotech Quality Framework (ICH M10-Aligned)
- Positive Reference Controls: Warfarin, propranolol, or target-specific benchmarks run alongside every batch
- Minimum n=3 Replicates: Independent measurements to establish precision and flag outliers
- Recovery Verification: Pre- and post-separation compound recovery must meet ≥80% acceptance threshold
- Blank Matrix Controls: System suitability and matrix effect assessment for every run
- Calibration Curves: R² ≥ 0.99, bracketing the concentration range of all study samples
What Causes Non-Specific Binding and Low Recovery — and How Do You Fix It?
Non-Specific Binding (NSB) happens when the compound sticks to non-target surfaces: tube plastics, filter membranes, assay plate walls. Lipophilic and basic compounds are especially prone. The result is artificially low free concentrations and an overestimate of true binding to protein.
Mitigation is concrete and well-validated:
Low-Binding Plastics
Polypropylene and coated low-adsorption consumables dramatically reduce passive compound loss before the assay measurement is even taken.
Surfactant Additives
Tween-20 at 0.01–0.05% coats hydrophobic plastic surfaces, blocking adsorption sites before the compound contacts the vessel wall.
Filter Pre-Conditioning
Blank matrix is passed through UF membranes before the study sample, saturating adsorption sites so the actual sample sees a pre-blocked surface.
“Calculating compound recovery pre- and post-separation is the single most important sanity check. If recovery drops below ~80%, the fu value is suspect — regardless of how clean the sensorgram looks.”
— Da-Ta Biotech Laboratory Protocol Standards
How Long Does a Protein Binding Assay Take?
Turnaround depends on whether the assay is off-the-shelf or custom. Standard high-throughput plasma protein binding screens (RED-based, with established LC-MS/MS) typically deliver in 2 to 5 business days per compound batch. Custom assays — novel targets, unusual matrices, or non-standard analytical methods — can stretch into weeks.
Key timeline drivers: target immobilization optimization (SPR/BLI), LC-MS/MS method development for proprietary compounds, validation depth requested (research-grade vs. GLP-aligned), and matrix complexity. Fast is good; rushed is expensive. Buffer development and validation steps protect data integrity downstream.
✅ Typical Turnaround Timeline at Da-Ta Biotech
- Standard PPB Screen (RED + LC-MS/MS): 2–5 business days
- SPR/BLI Target Binding (established target): 5–10 business days
- Custom Method Development (novel target/matrix): 2–6 weeks
- Full GLP-Aligned Validation Package: Timeline quoted per project scope
Why Does My SPR Experiment Show Binding in the Reference Channel?
Reference channel signal almost always means non-specific adsorption to the sensor matrix itself, not to your immobilized target. Left uncorrected, it inflates apparent binding and distorts kinetic fits.
Troubleshooting checklist: increase blocking agent concentration (BSA, casein, or carboxymethyl dextran derivatives); adjust running buffer pH and ionic strength; add low concentrations of detergent; verify reference surface activation matches the active channel. Proper double-referencing (subtracting both the reference flow cell and a blank injection) is essential — without it, kon and koff values will be wrong by a margin that no statistical correction can rescue.
Does High-Affinity Binding Present Assay Design Challenges?
When KD drops into the low picomolar range, standard assay formats hit physical limits. Target concentrations required for accurate measurement can fall below the sensor’s detection threshold, while koff becomes so slow that dissociation phases extend beyond practical run times.
Solutions include slower flow rates to capture extended dissociation, tracer displacement assays where a labeled competitor reports indirectly on binding occupancy, and extended measurement windows (sometimes hours per cycle). For very slow koff, single-cycle kinetics or pre-equilibrium analysis methods can recover accurate rate constants without waiting for full dissociation.
Can You Run Binding Assays in Complex Matrices Like Serum or Plasma?
Yes — but with method-appropriate caution. Real-time biosensor chips are vulnerable to bio-fouling from serum proteins and lipids, which can mask true binding signals or generate drifting baselines. SPR/BLI in raw plasma is possible but requires aggressive surface chemistry and short contact times.
For most plasma binding questions, the cleaner route is equilibrium dialysis or ultrafiltration followed by LC-MS/MS quantification. This separation-first approach bypasses matrix interference entirely: the binding equilibrium is established in physiological conditions, and the analytical readout happens in a clean buffer. Complex physiological matrices demand targeted separation strategies — they reward planning and punish improvisation.
“Complex physiological matrices demand targeted separation strategies — they reward planning and punish improvisation. Know your matrix before you design your assay.”
— Da-Ta Biotech Scientific Advisory Team
Frequently Asked Questions
Ready to Design Your Binding Study?
Which binding profile is currently blocking your lead optimization decision — affinity, kinetics, or plasma fu? Bring your scientific question, and the assay design will follow. Come to us with any binding challenge — WE ARE HERE FOR YOU.

