
In Vitro Pharmacology Assays: A Practical Guide for Drug Discovery R&D
Written by Dr. Rinat Borenshtain-Koreh, PhD, DVM — CEO of Da-Ta Biotech LTD, with over 25 years of hands-on experience in Biotech and Biomed R&D. Her teams have designed and validated hundreds of in vitro pharmacology assays supporting projects from early-stage proof-of-concept through FDA application packages.
Core claim: The assays you run in week two of a program determine whether you succeed or fail in year three. Rigorous in vitro pharmacology is not overhead — it is the most cost-effective investment in your pipeline.
🔬 Expert Insight
In vitro pharmacology assays are the working bench between a molecular idea and a biological reality. They measure how a drug candidate engages its target, how potent that engagement is, and whether the response is selective enough to justify further investment. For biotech and biomed teams, these assays provide the quantitative evidence that turns a chemical structure into a credible therapeutic hypothesis — and at Da-Ta Biotech, we treat pharmacological testing as a decision-making instrument, not a box-checking exercise.
Early data on potency, efficacy, and selectivity allow teams to triage molecules quickly — keeping the strong, retiring the weak, and avoiding costly late-stage failures. Done well, professional in vitro pharmacology assays serve as the foundation of the entire In Vitro Assays Drug Discovery pipeline, accelerating R&D solutions before any in vivo or clinical step is considered.
Regulators and developers alike treat in vitro characterization as mandatory before candidate molecules can move toward human testing. Profiling druggability properly — binding, function, ADME-related behavior, and off-target risk — is, simply put, the correct thing to do. For a broader context, see this overview on in vitro assay development.
What Are the Main Differences Between Biochemical and Cell-Based Assays?
Biochemical assays and cell-based assays answer different scientific questions. Biochemical formats focus on isolated proteins or enzymes — typically simpler, well-defined, and amenable to high-throughput screening. They are excellent for measuring direct binding, enzyme inhibition, or competition with a known ligand, where the readout is mechanistically clean.
Cell-based formats place the molecule into a living cellular environment. The readout reflects permeability, intracellular distribution, metabolic interactions, and pathway crosstalk — closer to physiology, but more complex to interpret. Scientists analyze drug effects on cells in controlled conditions using validated models such as those described in this In Vitro Cell Based Assay overview.
How to Select the Right Assay for Your Mechanism of Action (MOA)?
Assay selection starts from the biological hypothesis, not from the available platform. The target class — GPCRs, kinases, ion channels, nuclear receptors, or protein–protein interactions — dictates which readouts are mechanistically meaningful. A kinase program may rely on enzymatic activity and downstream phosphorylation, while a GPCR program may require second messenger signaling such as cAMP or calcium flux.
The desired pharmacological effect must align with the readout. Measuring gene expression when the mechanism is acute receptor activation will miss the signal. Choosing the wrong model — for example, ignoring intestinal permeability when oral absorption is critical — undermines “fit-for-purpose” results.
💡 Expert Methodology Note
Robust drug pharmacology services begin with a written assay rationale that ties the target, hypothesis, and readout together before any plate is run — ensuring that pharmacological testing delivers data the team can actually act on. At Da-Ta Biotech, this rationale document is a mandatory deliverable before project initiation.
- GPCR targets: cAMP accumulation, calcium flux, beta-arrestin recruitment
- Kinase targets: Enzymatic phosphorylation, HTRF-based detection, cell viability
- Ion channels: Patch-clamp electrophysiology, fluorescent dye-based flux assays
- Nuclear receptors: Reporter gene assays, TR-FRET coactivator recruitment
- PPI targets: AlphaScreen, SPR, label-free biophysical methods
Why Is an Assay Cascade Essential for Maximizing Drug Discovery Success?
An assay cascade is a structured sequence of tests designed to filter and optimize lead compounds. Rather than running every test on every molecule, the cascade routes compounds through progressively more demanding and more expensive assays. Hits that survive each stage carry stronger evidence of true activity.
This staged approach protects both budget and timeline. It also clarifies decision criteria: each tier has its own pass/fail thresholds for potency, selectivity, and quality metrics, making it easier to defend go/no-go calls in in vitro pharmacology programs.
🎯 Primary Assays
Primary assays prioritize speed and coverage — high-throughput screens that identify “hits” from large compound libraries. They are designed for maximum throughput with validated quality controls that ensure results are reliable before any secondary investment is made.
📈 Secondary Assays
Secondary assays validate functional activity, confirm specificity, and begin to characterize potency in more biologically relevant formats. Together they form the backbone of credible in vitro pharmacology assays, turning raw hits into actionable leads.
▶ Orthogonal Confirmation
Orthogonal assays use a different readout to answer the same biological question — for example, confirming a fluorescent-based hit with a label-free or radiometric method. This is the most reliable way to eliminate false positives driven by detection chemistry rather than real biology.
What Is Off-Target Screening and Why Is It Mandatory for Safety?
Off-target screening, also known as secondary pharmacology, tests a candidate against panels of receptors, transporters, enzymes, and ion channels that are not its intended target. The goal is to map unintended interactions before they appear as adverse effects in animals or patients.
This de-risking step has direct translational value: it can flag cardiovascular liabilities (such as hERG), CNS effects, or metabolic interferences early enough to redesign the molecule. Regulatory agencies recognize that pharmacological testing of secondary targets often correlates with clinical findings, which is why most modern drug pharmacology services include a defined off-target panel as part of standard lead profiling.
“The translational value of secondary pharmacology assays in predicting nonclinical and clinical findings cannot be overstated — these panels are now considered a regulatory expectation, not an optional add-on.”
— FDA Science Forum on Secondary Pharmacology Assays
⚠ Key Safety Flags to Screen
- hERG channel: Cardiac QT prolongation risk
- CNS receptor panels: Neurotoxicity and behavioral liabilities
- CYP enzyme inhibition: Drug–drug interaction potential
- Transporter panels: P-gp, BCRP, OATP-mediated interactions
Further reading on this is available from the FDA Science Forum on secondary pharmacology assays.
Common Mistakes That Produce False Positives in Assay Interference
False positives are the silent budget killer in screening campaigns. Pan-Assay Interference Compounds (PAINS), autofluorescent molecules, aggregators, and reactive species can all produce signals that look pharmacological but are purely artifactual. Without controls in place, an entire hit list can be contaminated.
⚠ PAINS Compounds
Reactive or promiscuous chemicals that interfere with assay detection chemistry rather than engaging the biological target specifically.
🔮 Aggregators
Colloidal aggregates sequester proteins non-specifically. Detergent addition (e.g., Triton X-100) in the assay buffer is the standard mitigation strategy.
💡 Autofluorescence
Compounds that absorb or emit at the same wavelength as the assay readout. Counterscreens at multiple wavelengths rapidly identify these optical artifacts.
Mitigation strategies are well-established: detergent in the assay buffer, counterscreens that detect optical interference, dose–response confirmation, and orthogonal readouts. As emphasized in Nature Reviews Chemistry, maintaining data integrity requires identifying chemical interference such as reactivity or fluorescence early. Embedding these controls into routine in vitro pharmacology assays is what separates a screening campaign that produces leads from one that produces noise.
What Does Professional Validation of In Vitro Pharmacology Assays Look Like?
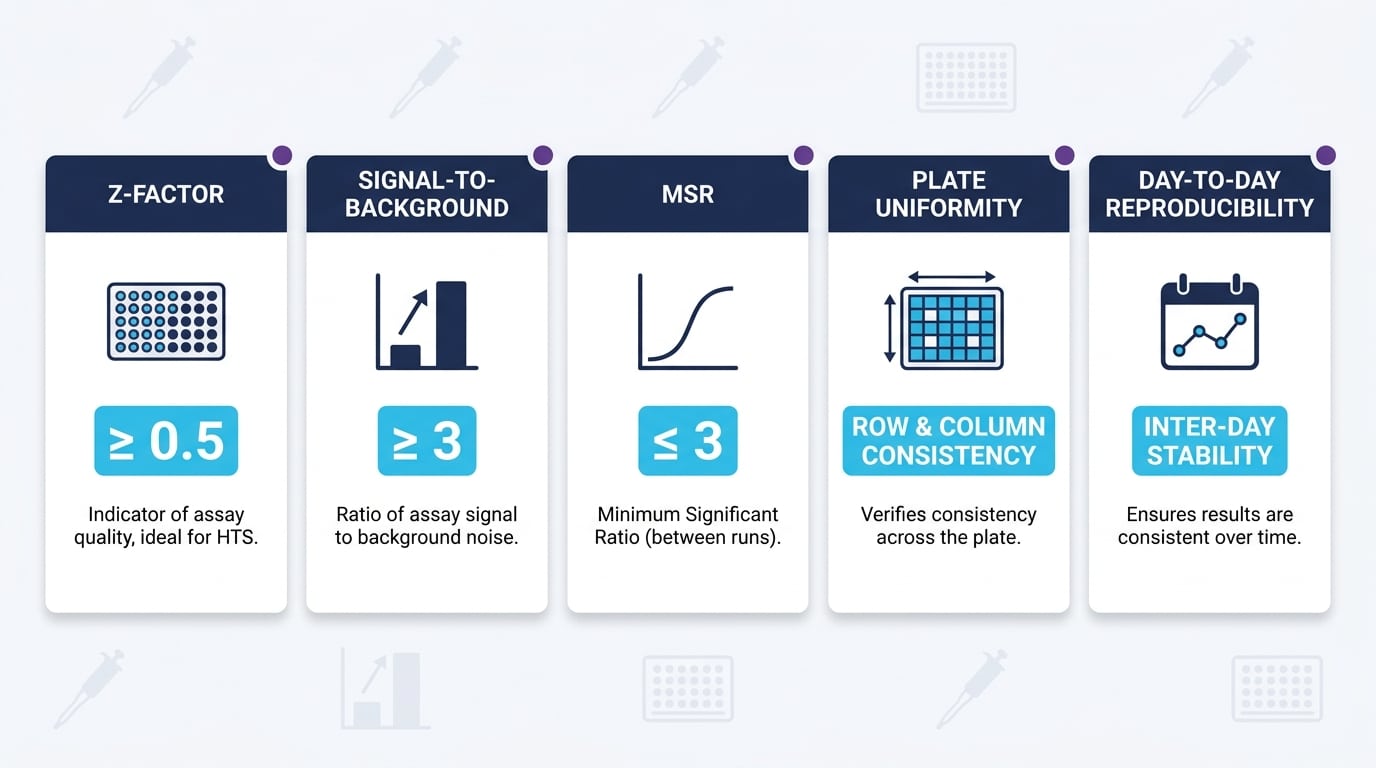
Validation is the bridge between a working protocol and a decision-grade assay. Key metrics define whether the assay is robust enough to support compound ranking and progression.
✓ Validation Best Practice
Plate uniformity and day-to-day reproducibility are not “nice to have” — they determine whether a 2-fold potency difference between two molecules is meaningful or noise. In rigorous in vitro pharmacology, these metrics are documented before a single screening plate is read, and they form a core deliverable of mature drug pharmacology services at Da-Ta Biotech.
How to Interpret IC50, EC50, and Dose-Response Curves?
IC50 is the concentration of a compound needed to inhibit a target response by 50%; EC50 is the concentration that produces 50% of maximal activation. Both values are derived from dose–response curves and are only as good as the curve fit and the underlying assay quality.
The Hill slope matters as much as the inflection point. A slope near 1 suggests a single, specific binding interaction; a steep slope may indicate cooperativity or non-specific mechanisms such as aggregation; a very shallow slope often points to heterogeneous binding.
Hill Slope ~1
Single, specific binding interaction. Ideal response indicating clean mechanism and appropriate assay conditions.
Hill Slope >2
Possible cooperativity, aggregation, or non-specific mechanism. Orthogonal confirmation urgently recommended.
Hill Slope <0.5
Heterogeneous binding, mixed populations, or assay artifact. Warrants mechanistic investigation before progression.
Statistically sound determination of IC50/EC50 — including appropriate weighting and inclusion of control data — is essential, as discussed in the Journal of Medicinal Chemistry. Without this discipline, pharmacological testing outputs become numbers without meaning.
“A concentration-response curve without proper controls, sufficient data points, and a documented Hill slope is a number masquerading as data. Decision-grade potency requires statistical rigor at every step.”
— Dr. Rinat Borenshtain-Koreh, CEO, Da-Ta Biotech LTD
Scenario: When an “Active” Compound Turns Out to Be an Artifact
📋 Case Study: The Autofluorescent Hit
Situation: A small-molecule program returns a confident primary screen hit with an IC50 of 1 µM — well within the activity threshold for progression to secondary testing.
Finding: In the secondary functional assay, potency disappears completely. A counterscreen reveals autofluorescence at the detection wavelength used in the primary format.
Impact avoided: Without the counterscreen, the team would have spent months — and significant budget — optimizing a chemical series with no real target engagement whatsoever.
Lesson: Orthogonal readouts, counterscreens, and validated controls are not optional add-ons. They are the difference between data you can defend and data that wastes a program. This is a core principle at Da-Ta Biotech.
🔬 Structural Recommendation
This is not a rare scenario — it is the everyday reality of screening. Embedding orthogonal confirmation into your assay cascade as a mandatory gate, not an optional follow-up, is the single highest-ROI structural change most drug discovery programs can make to their workflows.
What Deliverables Should You Expect from Drug Pharmacology Services?
Deliverables determine whether the data can travel — into the next experiment, into a partnership discussion, or into a regulatory submission. Standard outputs from professional drug pharmacology services include comprehensive study reports, raw data files, fitted dose–response graphs, and detailed SOPs and protocols.
🔌 Da-Ta Biotech Methodology
Every project at Da-Ta Biotech follows a documented four-phase workflow:
- 1Scientific scoping: Written assay rationale linking target, hypothesis, and readout
- 2Protocol development: Method development, counterscreens, and interference controls built in
- 3Validation gate: Z’-factor, S/B, MSR, and uniformity documented before any screening run
- 4Regulatory-ready reporting: Structured reports, raw files, and SOPs formatted for IND-style packages
For programs heading toward registration, the Israeli Ministry of Health requires detailed pre-clinical data as part of the new drug registration process. Well-structured in vitro pharmacology assays reports from Da-Ta Biotech are written to slot directly into IND-style packages, saving rework later.
Frequently Asked Questions
Ready to Design the Right Assay for Your Molecule?
What is the single scientific question your next assay must answer — and is your current protocol actually built to answer it? If you are planning a screening cascade, validating a lead series, or preparing pre-clinical packages for regulators, talk to a team that lives in this work every day.
Come to us with any scientific challenge — Da-Ta Biotech is here to help you profile the druggability of your molecule and move your program forward with confidence.

